Key Points
Rates of thrombosis and progression were low in patients with ET/PV treated with either HU or IFN in this randomized study.
PEG was more effective in normalizing counts and reducing JAK2V617F VAF in PV whereas HU induced more HPRs in ET.

Abstract
The goal of therapy for patients with essential thrombocythemia (ET) and polycythemia vera (PV) is to reduce thrombotic events by normalizing blood counts. Hydroxyurea (HU) and interferon-α (IFN-α) are the most frequently used cytoreductive options for patients with ET and PV at high risk for vascular complications. Myeloproliferative Disorders Research Consortium 112 was an investigator-initiated, phase 3 trial comparing HU to pegylated IFN-α (PEG) in treatment-naïve, high-risk patients with ET/PV. The primary endpoint was complete response (CR) rate at 12 months. A total of 168 patients were treated for a median of 81.0 weeks. CR for HU was 37% and 35% for PEG (P = .80) at 12 months. At 24 to 36 months, CR was 20% to 17% for HU and 29% to 33% for PEG. PEG led to a greater reduction in JAK2V617F at 24 months, but histopathologic responses were more frequent with HU. Thrombotic events and disease progression were infrequent in both arms, whereas grade 3/4 adverse events were more frequent with PEG (46% vs 28%). At 12 months of treatment, there was no significant difference in CR rates between HU and PEG. This study indicates that PEG and HU are both effective treatments for PV and ET. With longer treatment, PEG was more effective in normalizing blood counts and reducing driver mutation burden, whereas HU produced more histopathologic responses. Despite these differences, both agents did not differ in limiting thrombotic events and disease progression in high-risk patients with ET/PV. This trial was registered at www.clinicaltrials.gov as #NCT01259856.
Introduction
Polycythemia vera (PV) and essential thrombocythemia (ET) are myeloproliferative neoplasms (MPNs) characterized by specific driver mutations.1,2 ET and PV each have chronic courses complicated by thrombohemorrhagic events, systemic symptoms, splenomegaly, and progression to myelofibrosis (MF) and/or blast phase.3,4
Many drugs have been used to normalize the blood counts of high-risk patients with PV and ET. Although frequently used, the superiority of either interferon-α (IFN-α) or hydroxyurea (HU) has not been established.5 The Myeloproliferative Disorders Research Consortium (MPD-RC), therefore, conducted an independent, global, randomized, phase 3 trial of therapy-naïve, high-risk patients with ET/PV treated with peg-rIFN-α2a (PEG) or HU.
Patients and methods
MPD-RC 112 (NCT01259856) was conducted at 24 North American and European sites. The study was approved by the institutional review boards/ethical committees, informed consent was obtained from each patient prior to enrollment, and the trial was conducted in accordance with the Declaration of Helsinki. Eligibility required a diagnosis of ET or PV according to World Health Organization 2008 diagnostic criteria.6 High-risk disease was defined by 1 of the following factors: history of thrombosis, age >60 years, history of bleeding (ET only), platelet count >1500 × 109/L in ET and >1000 × 109/L in PV, vasomotor symptoms (erythromelalgia, severe migraine headaches), significant or symptomatic splenomegaly, and the presence of diabetes or hypertension requiring pharmacologic intervention (see supplemental Appendix for full inclusion/exclusion criteria; available on the Blood Web site). Therefore, in addition to classic risk factors for thrombohemorrhagic complications, patients were also included in this study if cytoreductive therapy was merited to address significant symptom and spleen burden.
Patients were randomly assigned 1:1 to HU or PEG within strata defined by disease type (ET/PV) using block randomization. PEG (provided by Roche/Genentech Pharmaceuticals, Nutley, NJ) was self-administered by subcutaneous injection at 45 μg weekly and titrated in 45-μg increments monthly to a maximum of 180 μg weekly. HU was initiated at 500 mg twice daily. Dose modification occurred when criteria for hematologic complete response (CR) were not met or dose-limiting toxicity occurred. Guidance and direction for dosing and escalations for both agents were provided in the protocol; the dosing protocol guidelines are included in the supplemental Appendix.
An intention-to-treat (ITT) evaluation was performed at 12, 24, and 36 months. Patients achieving at least a hematologic partial response (PR) remained on treatment, and length of therapy was determined by time of entrance into the study. The last subject to enter the study could receive therapy for at most 12 months, and the first subjects could receive therapy for up to 64 months. All patients were included in response assessments. The primary endpoint was the comparison of CR rates (as defined by European LeukemiaNet [ELN] criteria to after 12 months of therapy).7 CR was defined as a platelet count <400 × 109/L, hematocrit <45% without phlebotomy for patients with PV only, white blood cell count <10 × 109/L, resolution of splenomegaly, and resolution of disease-related symptoms (microvascular disturbances, headache, and pruritus). Responses were assessed by a central review committee blinded to treatment. Splenomegaly was assessed by ultrasound measurement of the craniocaudal axis (splenomegaly defined as >13 cm). Patients completed the Myeloproliferative Neoplasm Symptom Assessment Form, the European Organization for the Research and Treatment of Cancer Quality of Life Questionnaire-C30, and 5 exploratory questions to assess PEG-related side effects on a serial basis at baseline and 3, 6, 9, and 12 months.
Disease biomarkers
Bone marrow histopathology, karyotype, and myeloid gene mutation analyses were performed at baseline, after 12 and 24 months, and at study end. Histomorphologic remission was defined according to ELN and the revised ELN-IWG criteria and determined by blinded central expert review.7 Next-generation sequencing was performed using a targeted-sequencing panel designed to capture 156 genes implicated in myeloid malignancies, as previously described.8 Next-generation sequencing was performed on available samples (source of DNA was from peripheral blood granulocytes) at baseline, at 12, 24, and 48 months, and at study end. Samples were sequenced on Illumina HiSeq 4000 (Illumina, San Diego, CA) with an average target depth of 992× (126-bp paired-end reads).
Statistical analysis
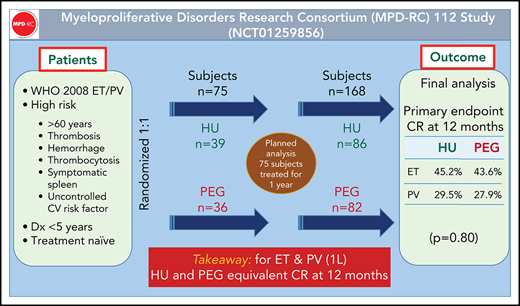
The study was designed to include 300 patients to detect a difference in complete clinical hematologic response rate of 15% from 15% (assumed HU rate) to 30% (assumed PEG rate, α = 0.05, two-sided, power = 0.85). In 2013, the study was amended from an original sample size of 612 total patients (306 per ET/PV strata) to allow increased disease duration from 3 to 5 years. Under the amended protocol, planned interim analysis was conducted in July 2016 after the first 75 patients were enrolled in the study (HU: 39; PEG: 36). The CR rate for HU was 33% (13/39) and 28% (10/36) for PEG (P = .60), which did not cross stopping boundaries for either efficacy or futility. The overall response rate (ORR; CR + PR) was 69% for HU (27/39) and 81% (29/36) for PEG. The study was continued until enrollment reached 168 patients, when a decision was made by the manufacturer to halt PEG access. With 170 patients randomized to 2 treatment groups in 2 strata, a difference of 19% in CR rates (from 15% to 34%) could be detected for the 2 disease strata combined, with a power of 82%.
Patients who dropped out due to lack of response, tolerability, or complications before the 12-month evaluation were considered nonresponders per ITT analysis. For response assessment at 24 months, only patients enrolled before June 2015 were included (n = 106 due to study closure) and analyzed in an ITT fashion. Similarly, for response assessment at 36 months, only patients enrolled before June 2014 were evaluated.
The primary analysis consisted of a comparison of proportions for CR and ORR by arm based on a 2-sided z-test for proportions. The Cochran-Mantel-Haenszel test for heterogeneity for disease strata (ET/PV) was also conducted. ET and PV rates are reported separately, but all comparisons were conducted for patients with combined ET/PV by arm only. Safety and toxicity were summarized to compare the distributions of incidence and severity of adverse events (AEs) by treatment. Patient-reported outcome measurements were scored according to published scoring algorithms. Baseline scores were compared between treatment arms with a 2-sided 2-sample independent sample Student t test. Overall complication-free survival was defined as free of major thrombotic event, major hemorrhagic complications, progression to MF, progression to acute leukemia, or death and was compared using a 2-sided log-rank test of the hazard ratio of PEG as compared with patients treated with HU. Exploratory analysis for mutational status at baseline and association with obtaining CR was analyzed by multivariable logistic regression. Variables included in the model were treatment, disease type, JAK2, CALR, ASXL1, and TET2 mutational status. JAK2V617F variant allele frequency (VAF) at baseline was analyzed according to response status by Wilcoxon rank-sum test. In addition, JAK2V617F VAF over time was evaluated using a linear mixed model. Time (months), treatment, and a treatment-by-time interaction were included as fixed effects, while an individual-specific random effect was included. SAS version 9.4 (SAS Institute, Cary, NC) was used for analysis, and values of P < .05 (2-sided) were considered statistically significant.
Results
Patients
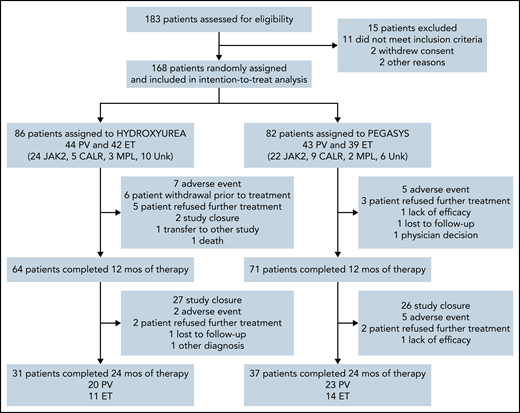
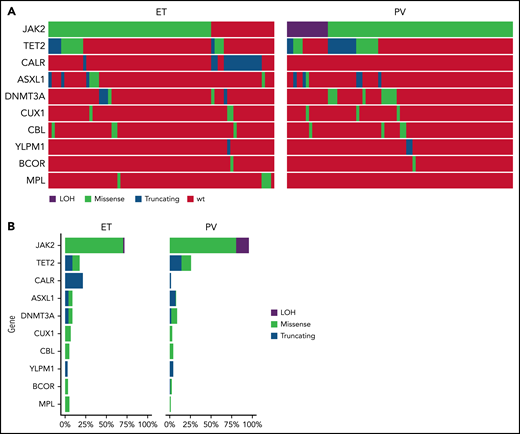
A total of 168 patients were enrolled from September 2011 to June 2016; 86 were randomized to HU and 82 to PEG (Figure 1). Eighty-one patients had ET and 87 had PV; baseline demographics are shown in Table 1. Risk factor profile for enrolled patients indicated that 82% of patients with PV and 75% of patients with ET were high risk due to age ≥60 years and/or a history of thrombosis. The risk profile for the remaining 16 patients with PV and 20 patients with ET is shown in supplemental Table 1. Among MPN driver mutations, JAK2V617F were most frequent (91%), followed by CALR (8%) and MPL mutations (3%). The most frequent co-occurring mutations involved TET2 (24%) and ASXL1 (9%) (Figure 2). Median JAK2V617F VAFs at baseline were 13% and 35% in patients with ET and PV, respectively. Baseline MPN symptoms and quality of life scores are shown in supplemental Table 2 and supplemental Figure 1.
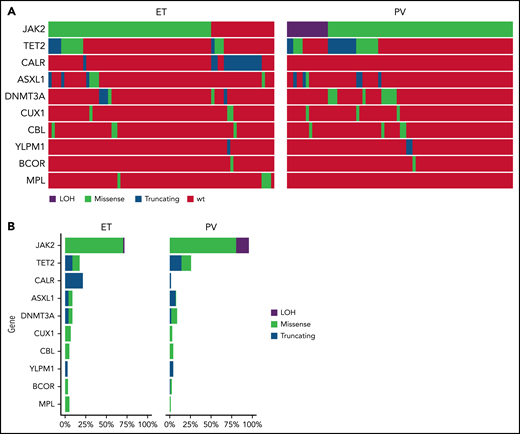
Mutational analysis of patients treated in MPD-RC 112 trial. (A) Oncoprint of baseline mutations and (B) mutation frequency of patients enrolled in the study.
Mutational analysis of patients treated in MPD-RC 112 trial. (A) Oncoprint of baseline mutations and (B) mutation frequency of patients enrolled in the study.
Responses
Clinicohematologic response
All 168 patients were included in response assessment per ITT analysis at 12 months. Six patients in the HU treatment arm withdrew before starting treatment; all patients in the PEG arm received treatment. The median (range) duration of treatment was 81.0 weeks (0-268) and 94.6 weeks (2.9-287.3) in the HU and PEG arms, respectively. Median weekly dose of HU was 6708 mg and 89.4 μg for PEG. Seventy-four percent of patients receiving HU and 87% of patients receiving PEG were treated for 12 months or longer.
CR at 12 months was 37% for HU and 35% for PEG (P = .80). ORR at 12 months was 70% for HU and 78% for PEG (P = .22; Table 2). In patients with ET, CR at 12 months was observed in 45% of those receiving HU and 44% for those receiving PEG. In patients with PV, CR at 12 months was observed in 30% for those receiving HU and 28% for those receiving PEG. ORR at 12 months was 70% for HU and 78% for PEG (P = .22; Table 2). Sensitivity analysis (accounting for 6 patients on HU who withdrew before treatment) for CR at 12 months for HU arm was 40% (difference of 5%; 95% CI, −11% to 20% vs PEG) and ORR was 75%.
CR at 12 months (without spleen normalization) was seen in 42% of patients receiving HU and 48% of patients receiving PEG (difference of −6% [95% CI, −9 to 21]). Forty-three percent and 65% of patients with PV receiving HU and PEG, respectively, achieved hematocrit control (without phlebotomy) at 12 months (P = .04; supplemental Figure 2). Platelet control in patients with ET at 12 months was 45% and 46% for patients receiving HU and PEG, respectively (P = .93). Alleviation of symptoms was the criterion least frequently met for achieving a CR, and when not requiring symptom resolution, there were no notable differences between CR rates by treatment arm. In patients with PV, 41/87 (HU: 21; PEG: 20; 47%) were receiving phlebotomy during the prior 6 months before enrollment, with a median number of phlebotomies of 3.0 (1-12) for HU and 4.0 (1-13) for PEG. Thirty-three patients with PV (HU: 15; PEG: 18) received phlebotomy during the first year, with a median number of phlebotomies of 2.0 (1-5) for HU and 3.0 (1-6) for PEG.
Median weekly dose in patients with CR (during first 12 months) as compared with patients without CR was 6995 mg (interquartile range [IQR]: 5567-7678) and 5804 mg (IQR: 3884-7000) for HU (P = .03). Median weekly dose for PEG was 76.4 μg (IQR: 46.7-104.4) for patients with CR and 89.2 μg (IQR: 59.7-131.2) for patients without CR (P = .27; supplemental Figure 3).
In 106 patients (HU: 54; PEG: 52) per ITT analysis, the CR at 24 months was 20% for HU and 29% for PEG, and ORR was 41% for HU and 60% for PEG (P = .045 for ORR comparison). In 57 patients (HU: 30; PEG: 27), the CR at 36 months was 177% for HU and 33% for PEG, and ORR was 47% for HU and 59% for PEG (P = .34 for ORR comparison). Twelve-month rates for the 24- and 36-month subgroups were similar to those reported for the full analysis dataset.
Spleen response
One hundred nine patients (HU: 51; PEG: 58) received post-baseline imaging for spleen response. Median spleen reduction (best response on treatment) was −5% (−24% to 17%) in patients treated with HU compared with a median reduction of −6% (−37% to 54%) with PEG (supplemental Figure 4). In patients with a spleen size ≥13 cm by imaging at baseline, 4/37 (11%) receiving HU attained a normalized spleen compared with 6/36 (17%) receiving PEG at any time on treatment.
Bone marrow response
In 109 evaluable patients (pre- and posttreatment biopsy), HU had a histopathologic response (HPR) of 12/52 (23%) vs 3/57 (5%) for PEG (P = .01; supplemental Figure 5). Best HPR response was seen in 18/54 (33%) of patients receiving HU and 10/59 (17%) patients receiving PEG (P = .05). HPR was more frequent in the ET cohort at 12 months 13/46 (24%) as well as best response 20/48 (42%) compared with the PV cohort at 4/63 (6%) and 8/66 (12%), respectively. A dose-dependent effect on achieving HPR was observed with HU (P = .04) but was not observed with PEG treatment (P = .6; supplemental Figure 6).
Cytogenetic response
One hundred forty-four of 168 (86%) patients had baseline cytogenetics. Abnormalities were seen in 22/144 (15%) patients: 14/71 (20%) patients treated with HU and 8/73 (11%) patients treated with PEG. Three of 14 (21%) patients treated with HU and 3/8 (38%) patients treated with PEG had a chromosomal abnormality [trisomy 9, del(20q), t(14;21)(q24;q22); trisomy 8, and loss of Y chromosome] (P = .62). After 24 to 36 months of follow-up, 3/71 (4%) patients receiving HU and 1/73 (1%) patients receiving PEG had gain of del(20q), loss of Y and del(16q) (supplemental Table 3). There was no association between cytogenetic response and CR/PR.
Molecular response
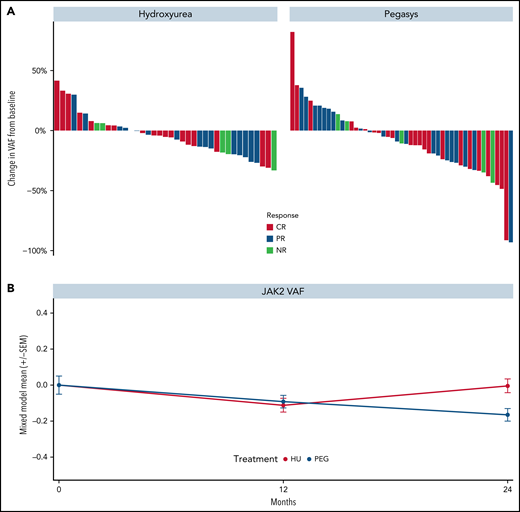
Reductions in JAK2V617F VAF were observed in most patients treated with HU or PEG (Figure 3A-B, respectively) and no differences were seen by clinical response. Results were similar for ET and PV groups (supplemental Figure 7A). Similarly, decreases in CALR and TET2 VAFs were observed in both treatment arms (supplemental Figure 8A-B, respectively). The median greatest change from baseline in JAK2V617F VAF was −5.3% and −10.7% in patients treated with HU and PEG, respectively. Notably, the median JAK2V617F VAF decreased consistently from baseline through month 24 in the PEG arm but increased in the HU arm after month 12. Mixed-model estimates for JAK2 VAF reduction at 24 months were −0.004 (95% CI, −0.08, 0.08) for HU and −0.16 (95% CI, −0.23, −0.10) for PEG (P = .002 for interaction effect; Figure 3B). No significant differences were observed between baseline JAK2V617F VAF in responders and nonresponders treated with either drug (supplemental Figure 8C-D, respectively). The driver mutation type was not associated with achieving response. By contrast, an ASXL1 mutation was associated with decreased odds of achieving hematologic CR status on multivariable analysis (P = .055; supplemental Figure 8E).
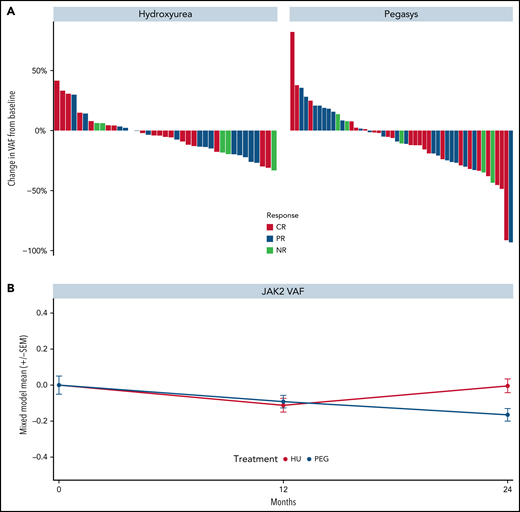
Kinetics of JAK2V617F allele burden of patients treated in the MPD-RC 112 trial. (A) Maximum change in JAK2V617F allele burden from baseline by response status. (B) JAK2V617F allele burden over time (*P < .05). Baseline (n = 117), 12-month (n = 97), and 24-month (n = 52) allele burden values were included in the mixed model.
Kinetics of JAK2V617F allele burden of patients treated in the MPD-RC 112 trial. (A) Maximum change in JAK2V617F allele burden from baseline by response status. (B) JAK2V617F allele burden over time (*P < .05). Baseline (n = 117), 12-month (n = 97), and 24-month (n = 52) allele burden values were included in the mixed model.
Complication-free survival
Five events (HU: 3; PEG: 2) occurred during the trial with an hazard ratio = 0.60 (95% CI, 0.10-3.62). One patient treated with PEG had a bleeding event consisting of macroscopic hematuria that required red cell transfusions. A second patient treated with PEG had a cerebral vascular accident. One patient treated with HU developed bilateral vertebral artery blockage noted on imaging but without clinical consequences. One patient treated with HU progressed to MF after 46 months. One patient treated with HU died of lung cancer at 9 months. Cumulative incidence of thrombosis was 2% (95% CI, 0.3-13) for HU and 2% (95% CI, 0.3-15) for PEG at 24 months.
Safety
The causes for therapy discontinuation are listed in supplemental Table 4. Discontinuation due to study closure occurred in 94 (56%) patients at a median of 25.4 (95% CI, 23.1-29.7) months.
One hundred sixty-two of 168 (96%) patients were assessed for Aes (HU: 80; PEG: 82). Grade 3 or higher (any attribution) Aes occurred in 60 (37%) patients (HU: 22 [28%]; PEG: 38 [46%]) (Table 3; z = −2.48; P = .01). PEG-related Aes were common (flu-like symptoms, injection site reaction, peripheral sensory neuropathy, blurred vision). Grade 1-2 depression was seen in 15% of patients receiving PEG as compared with 3% of patients receiving HU. Mucositis and anorexia were significantly more common in the HU arm, whereas leukopenia, flu-like symptoms, injection site reactions, aspartate aminotransferase elevations, and depression occurred more frequently in the PEG arm. In addition, hypertension, fatigue, and lymphopenia were higher in the PEG arm.
Discussion
The continued controversy over the choice of drug for first-line therapy for patients with ET or PV who are at an increased risk of developing thrombotic events9 is a consequence of the limited numbers of randomized clinical trials performed and the chronic nature of these malignancies limiting the number of thrombotic events that occur during follow-up.5,10
Our data indicate that PEG and HU are both effective treatments for PV and ET at 12 months without significant difference in CR. These findings are consistent with PEG therapy requiring greater time to achieve full clinical potential. PEG may be more effective in correcting blood counts in PV compared with ET because the percentage of patients with PV who achieved hematocrit control at 12 months was higher (65% vs 43%) whereas platelet control in patients with ET at 12 months was similar between arms (45% vs 46%). The median change in JAK2V617F VAF observed in patients treated with HU was −5.3% and −10.7% for PEG. Notably, the median JAK2V617F VAF decreased through month 48 with PEG but increased with HU after month 12. Because PEG is thought to act at the level of the mutated MPN stem cell,11-14 more frequent histological responses in patients treated with HU were unanticipated. Given the noted association between HU dose and rate of hematologic CR and bone marrow HPR, it is possible that HU dose-related myelosuppression may account for the higher HPR seen here. Furthermore, bone marrow histopathological responses occurred more commonly in patients with ET than PV although the clinicohematological ORR was higher in patients with PV. Cytogenetic abnormalities were lost in a similar number of patients receiving either drug. The discordance between the greater reduction of JAK2V617F VAF due to PEG and the improvement in marrow morphology with HU is difficult to reconcile.15
The number of thrombotic and progressive events in either arm was small, limiting the ability to detect differences in thrombosis between agents. Both agents were associated with few serious Aes. Mucositis and anorexia were more common in the HU group, whereas PEG was associated with Aes such as injection site reactions, flu-like symptoms, peripheral sensory neuropathy, leukopenia, depression, and alanine aminotransferase elevations.
Contemporaneously with this study, the PROUD/CONTINUATION-PV trial compared HU and ropeginterferon alfa-2b (Ro-PEG) in PV. In PROUD-PV, Ro-PEG was not inferior to HU after 12 months, with a CR rate of 21.3% vs 27.6% in data analyzed from 245/257 patients.16 In the CONTINUATION-PV trial, 171 patients continued therapy, and at 60 months, Ro-PEG and HU had CHR rates (control of blood counts and absence of phlebotomy) of 55.8% and 44.0% (P = .0974), respectively.17 A difference in the incidence of thrombotic events or evidence of progression was not observed. Similar to MPD-RC 112, the degree of reduction in JAK2V617F VAF persisted and progressively increased exclusively in the Ro-PEG arm. The PROUD/CONTINUATION-PV study did not include patients with ET, bone marrow histopathology, or cytogenetics or monitor the effects of these treatments on patient symptoms when evaluating CR, which may account for their higher CR rates.
Limitations to the MPD-RC 112 study existed. The trial was closed after the accrual of 168 patients due to lack of availability of PEG. The lower-than-expected accrual and limited follow-up impacted the ability to evaluate longer term outcomes and evaluate ET and PV strata separately. Based on accrual of 168 patients, conditional power was 12% for a targeted sample size of 300 patients (150 per strata). Had the study continued to full enrollment of 300 patients, there was a low likelihood of observing a superiority result with PEG. This study showcases the difficulty of performing clinical trials in high-risk patients with PV and ET, in whom the goal is ultimately to reduce thrombotic episodes or disease progression. Although the bone marrow HPRs are of interest, their evaluation by a single blinded expert hematopathologist without the use of a central review committee limits their impact. The starting dose and titration of each agent was outlined in the protocol and may differ from the variable real-world dosing approach of HU or PEG, which can affect both tolerability and potentially efficacy. Given the divergence of molecular response and bone marrow histopathologic remission, we raise caution of using changes in JAK2V617F VAF, marrow histopathology, or clearance of cytogenetic abnormalities as measures of disease course modification.
No difference in CR at 12 months was seen between treatment arms. However, after 24 months of treatment, a difference in ORR could be discerned between PEG and HU therapy, yet the incidence of thrombotic events or evolution to MF and MPN–blast phase did not differ. Grade ¾ treatment-emergent Aes were more frequent with PEG. We conclude that both HU and PEG are effective agents in normalizing blood counts and limiting the number of thrombotic events in patients with high-risk ET/PV, and the decision to choose 1 agent over the other must be personalized.
Acknowledgments
This work was supported by a grant from the National Cancer Institute, National Institutes of Health (MPN Research Consortium, 5P01CA108671-09), a Cancer Center Support Grant/Core grant to Memorial Sloan-Kettering Cancer Center (P30 CA008748), and generous independent, unrestricted support from Roche Genentech. R.K.R. is supported by the National Cancer Institute, National Institutes of Health (1K08CA188529-01).
Authorship
Contribution: J.M., H.E.K., J.T.P., A.R., C.N.H., M.F.M., A.M.V., J.E., J.-J.K., R.T.S., R.S.W., L.P., J.D.G., T.B., R.M., G.T., R.A.M., A.C.D., and R.H. contributed to the design of the clinical trial; J.M., H.E.K., A.R., D.B., A.Y., C.N.H., M.F.M., A.M.V., J.E., C.L.O., J.-J.K., A.J.M., E.F.W., D.S.L., V.D.S., M.O.A., C.M.K., R.C., D.R., R.T.S., A.B., A.N., M.K., M.E.S., V.N., N.F., R.S.W., L.P., J.D.G., T.B., R.M., G.T., R.K.R., R.A.M., A.C.D., and R.H. wrote the manuscript; J.M., H.E.K., J.T.P., A.R., D.B., A.Y., C.N.H., M.F.M., A.M.V., J.E., C.L.O., J.-J.K., A.J.M., E.F.W., D.S.L., V.D.S., M.O.A., C.M.K., R.C., D.R., R.T.S., A.B., A.N., M.K., M.F.L., J.E.A.O., E.M., L.S., M.E.S., V.N., J.T., N.F., A.V.P., R.S.W., L.P., J.D.G., T.B., R.M., G.T., R.K.R., R.A.M., A.C.D., and R.H. contributed to patient recruitment, data collection, and analysis; H.E.K., L.P., J.D.G., and A.C.D. performed statistical analyses; and all authors gave final approval of the version to be published and agreed to be accountable for questions related to the accuracy and integrity of the work.
Conflict-of-interest disclosure: J.M. reports clinical trial research support paid to the institution from Incyte, Roche, Novartis, CTI Biopharma, Janssen Pharmaceuticals, Geron, Kartos, AbbVie, Merck, Promedior, PharmaEssentia, and Celgene/BMS and is a clinical trial steering committee member, scientific advisory board member, and consultant for Roche, CTI Biopharma, Constellation, Novartis, Galecto, PharmaEssentia, Sierra Oncology, AbbVie, Kartos, Incyte, and Celgene/BMS. reports clinical trial research support paid to the institution from AbbVie, AI, Astellas, Forma, Incyte, Kite, and Takeda. E.F.W. reports advisory boards for Incyte and Gilead Sciences. A.Y. reports consultation and speaker honoraria for Incyte, Seattle Genetics, and Novartis. A.R. reports consultation and speaker honoraria for Novartis, Amgen, Roche, Celgene, and Italfarmaco. A.M.V. reports speaker honoraria from Novartis and Celgene and fees for participation on advisory boards for Novartis, CTI, and Celgene. D.R. reports consulting honoraria from Incyte. M.O.A. reports research grant support paid to institution from Incyte, CTI Biopharma, Samus Therapeutics, Janssen Pharmaceuticals, and Gilead. R.T.S. reports consultancy and speaker bureau fees from Pharmaessentia. R.K.R. has received consulting fees from Stemline Corporation, Celgene, Agios Pharmaceuticals, Apexx Oncology, Beyond Spring, Partner Therapeutics, and Jazz Pharmaceuticals and has received research funding from Constellation Pharmaceuticals, Incyte, and Stemline Therapeutics. R.M. reports research support from Incyte, Genentech, CTI, Promedior, and AbbVie and is a consultant for Novartis, Sierra Oncology, and La Jolla Pharma. R.H. reports research support from Roche. The remaining authors declare no competing financial interests.
Correspondence: John Mascarenhas, Tisch Cancer Institute, Division of Hematology/Oncology, Icahn School of Medicine at Mount Sinai, One Gustave L. Levy Place, Box 1079, New York, NY 10029; e-mail: john.mascarenhas@mssm.edu.
Presented in abstract form the 2018 Annual Meeting of the American Society of Hematology, San Diego, CA, 1-4 December 2018.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.