

In this issue of Blood, Guo et al1 identify an essential role for the nuclear factor hemogen, also called erythroid differentiation-associated gene (EDAG), in erythroid maturation and GATA1-mediated gene activation.
Hemogen expression is normally restricted to the hematopoietic system, where it regulates proliferation, differentiation, and survival of hematopoietic cells.2 Elevated hemogen expression is associated with malignant transformation to either acute myeloid leukemia or acute lymphocytic leukemia.3 Conversely, decreased hemogen levels are associated with dyserythropoiesis in patients with myelodysplastic syndrome (MDS).4 Highlighting its clinical relevance, increasing the expression of hemogen improves erythroid differentiation of CD34+ cells from patients with MDS.4 Hemogen has been implicated in the regulation of erythropoiesis and is a direct target of the transcription factor GATA1.3 In addition, hemogen promotes GATA1 acetylation through recruitment of P3005 and regulates GATA1 stability through interaction with HSP70.4 Guo et al identified an essential role for hemogen in normal erythroid maturation by studying hemogen disruption in the erythroid HUDEP2 cell line and in primary human CD34+-derived erythroid cells, as well as in a murine knockout model. Transcriptomic analyses performed after hemogen disruption suggest that hemogen is important for GATA1-mediated gene activation, thereby having an impact on the expression of the globin genes and heme synthesis enzymes.
GATA1 modulates gene expression through interaction with co-activators such as LMO2 and LDB1 as well as co-repressors, including ETO2 and the nucleosome remodeling and deacetylase (NuRD) complex.6,7 These factors have antagonistic roles in chromatin structure, and the balance of their action ultimately regulates the level of GATA1-mediated gene expression. The question of how GATA1 “decides” which factors to recruit to a specific region has been intensely studied but remains incompletely understood. Guo et al shed light on this question by demonstrating that at the beta globin locus, hemogen loss leads to decreased occupancy of GATA1 and LDB1, inefficient chromatin looping, and increased occupancy of the factor ETO2 and the NuRD complex member CHD4. These results suggest that hemogen promotes formation of the activating LDB1 complex at the expense of recruiting ETO2 and the NuRD complex. The authors then identify hemogen-interacting proteins using mass spectrometry and co-immunoprecipitation experiments. They demonstrated that hemogen interacts with GATA1 and LDB1, as well as the cohesin complex, an important mediator of higher-order chromatin interactions, and the SWI/SNF chromatin remodeling complex, which regulates nucleosome positioning and DNA accessibility at regulatory elements such as enhancers and promoters. Consistent with these results, hemogen knockout in K562 cells leads to loss of SWI/SNF complex enrichment at the beta globin locus, accompanied by a loss of chroman accessibility and histone acetylation. The authors extend these findings in vivo, using a mouse model that expresses hemagglutinin (HA)-tagged hemogen to demonstrate that hemogen extensively co-localizes with the SWI/SNF complex member BRG1 and histone H3, lysine 27 acetylation (H3K27Ac), a mark of active enhancers and promoters. They further use their hemogen-null mouse model to demonstrate that hemogen disruption abrogates recruitment of BRG1 to target genes, leading to loss of chromatin accessibly at those regions. Loss of hemogen also resulted in lower H3K27Ac levels, consistent with loss of BRG1 and increased occupancy of the NuRD complex. Taken together, their data support a model in which hemogen recruits the SWI/SNF complex member BRG1 to GATA1 complexes and excludes ETO2 and the NuRD complex to promote erythroid-specific gene expression (see figure).
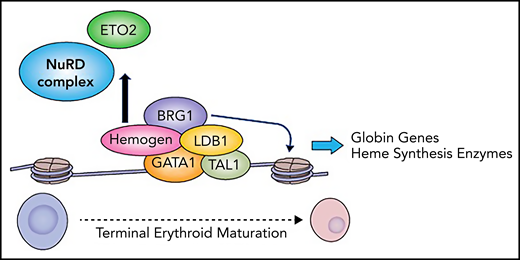
Hemogen regulates GATA1 complex composition. Hemogen interacts with GATA1, recruiting the SWI/SNF chromatin remodeler BRG1 to form the activating LDB1 complex and to exclude ETO2 and the NuRD complex, facilitating high-level erythroid gene expression during terminal erythroid maturation. Professional illustration by Patrick Lane, ScEYEnce Studios.
Hemogen regulates GATA1 complex composition. Hemogen interacts with GATA1, recruiting the SWI/SNF chromatin remodeler BRG1 to form the activating LDB1 complex and to exclude ETO2 and the NuRD complex, facilitating high-level erythroid gene expression during terminal erythroid maturation. Professional illustration by Patrick Lane, ScEYEnce Studios.
The synthesis of heme and globin are key priorities of red cell precursors, and the expression of these genes is driven by the master transcriptional regulator GATA1. In erythroid precursors, GATA1 operates in the context of a nucleus in which transcriptional co-repressors are vastly more abundant than co-activators,8 raising the fundamental question of how GATA1 recruits co-activators to drive expression of heme synthesis enzymes and other erythroid-specific genes. This question is clinically relevant because appropriate expression of these genes is essential for erythropoiesis, and failure to properly express these genes results in inherited and acquired anemias.9 The work of Guo et al suggests that hemogen is a novel player in GATA1-mediated gene activation that promotes interaction of GATA1 with co-activators at the expense of co-repressors. They further demonstrate that hemogen is important for the establishment of GATA1 complexes that promote appropriate patterns of histone acetylation, chromatin accessibility, and chromatin looping. Many of these activities are likely a result of the cooperativity of hemogen with BRG1. How hemogen is recruited to specific GATA1 sites and whether hemogen can interact with other transcriptional regulators remain open questions, and further study in this area will provide important insights into how transcription networks are established in maturing erythroid cells. Intriguingly, some of these interactions may be developmentally regulated. Hemogen-null mice have defects in fetal liver erythropoiesis; however, adult mice are viable without an obvious phenotype. Consistent with these findings, hemogen is highly expressed in murine fetal erythroid cells, but it is expressed at much lower levels in adult bone marrow.10 In contrast to the murine system, expression of hemogen seems to be similar in human fetal and adult hemopoietic progenitors, suggesting that there may be species-specific differences in the mechanisms that underlie GATA1-mediated gene activation. Further work is needed to understand the formation and function of these complexes, which will provide important insights into mechanisms underlying GATA1-mediated gene regulation and developmental hemoglobin switching. Ongoing studies in this area have the potential to provide important insights into the mechanisms underlying both inherited and acquired anemias and to reveal novel strategies for therapeutic intervention.
Conflict-of-interest disclosure: The author declares no competing financial interests.

