

In this issue of Blood, 2 independent studies, by Kimura et al1 and Passet et al,2 describe a novel genetic subtype of B-progenitor acute lymphoblastic leukemia (B-ALL) characterized by 2 separate focal genomic deletions, leading to enhancer “retargeting,” causing upregulation of CDX2 at chromosome 13, and to the formation of a chimeric gene fusion (UBTF::ATXN7L3) at chromosome 17. This novel subtype is rare, identified in ∼1% to 2% of B-ALL depending on the study cohorts. One might ask, Why even bother investigating such a rare subtype of ALL? The 2 studies show that this is worthwhile from both a biological and a clinical perspective.
B-ALL is caused by a vast number of recurrent somatically acquired genetic alterations of critical biological and clinical importance. These genetic alterations show us which genes are important in malignant hematopoiesis, many of which also play a critical role in normal B-cell development. By studying the altered genes and their effect on other genes/proteins and biological/cellular pathways, we can deduce how ALL arises, and it is hoped, also provide a basis for the development of new therapies. From a clinical perspective, the genetic alterations are of vital importance because they are used to establish a firm diagnosis, to risk-classify patients, to select treatment, and to monitor treatment effects.
Currently, the 2016 World Health Organization classification recognizes 7 genetic B-ALL entities and 2 provisional ones.3 With the introduction of whole-genome sequencing (WGS) and whole-transcriptome sequencing (WTS), several new subtypes (>15) of B-ALL have been identified,4-6 providing a better understanding of the “B-other ALL” group that historically lacked characteristic genetic changes. Many of these new subtypes are, unfortunately, not detectable by the current gold-standard methods in a clinical diagnostic setting, instead requiring the use of WGS and WTS. The fraction of B-other ALL decreases to between 5% and 8% of B-ALL cases depending on the age group (see figure panel A) when high-throughput sequencing techniques are used.4-6 Kimura et al and Passet et al now take a further step toward narrowing the gap of unknown genetic causes of B-other ALL.
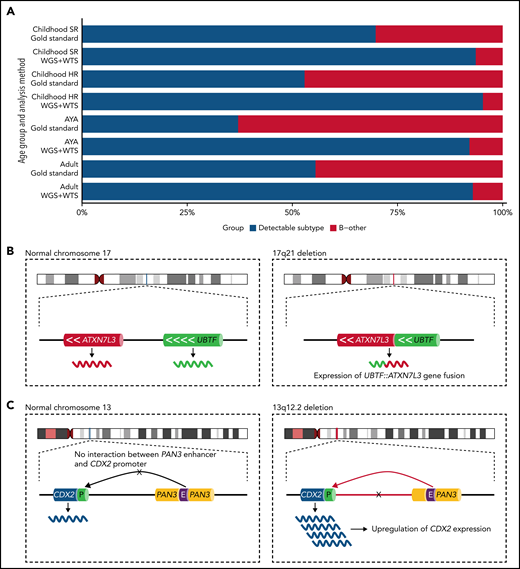
The frequency of B-other in different age groups of B-ALL and the genetic rearrangements characterizing the newly described UBTF/CDX2 ALL subtype. (A) An estimate of the fraction of B-ALL cases harboring a subtype-specific genetic alteration and those with an unknown genetic cause (B-other) when assessed by gold-standard methods or WGS and WTS. Gold-standard diagnostics typically include chromosome banding, fluorescence in situ hybridization (FISH), and targeted molecular tests. About 20% of the Philadelphia-like B-ALL subtype was estimated to be detectable indirectly by FISH. Frequency estimates were inferred from Gu et al and Kimura et al.5,6 (B, left) The localization of the UBTF and ATXNL3 genes at 17q21 are schematically depicted. (right) A focal 10-kb genomic results in an in-frame UBTF::ATXN7L3 gene fusion at the breakpoint junction. (C, left) Schematic depiction of 13q12.2 in which the CDX2 gene is located ∼280 kb away from the active PAN3 gene enhancer. (right) A focal genomic deletion causes an aberrant chromatin looping configuration by which an active enhancer of the PAN3 gene is retargeted (“hijacked”) and starts driving the ectopic expression of the CDX2 gene. AYA, adolescents and young adults; HR, high risk; SR, standard risk.
The frequency of B-other in different age groups of B-ALL and the genetic rearrangements characterizing the newly described UBTF/CDX2 ALL subtype. (A) An estimate of the fraction of B-ALL cases harboring a subtype-specific genetic alteration and those with an unknown genetic cause (B-other) when assessed by gold-standard methods or WGS and WTS. Gold-standard diagnostics typically include chromosome banding, fluorescence in situ hybridization (FISH), and targeted molecular tests. About 20% of the Philadelphia-like B-ALL subtype was estimated to be detectable indirectly by FISH. Frequency estimates were inferred from Gu et al and Kimura et al.5,6 (B, left) The localization of the UBTF and ATXNL3 genes at 17q21 are schematically depicted. (right) A focal 10-kb genomic results in an in-frame UBTF::ATXN7L3 gene fusion at the breakpoint junction. (C, left) Schematic depiction of 13q12.2 in which the CDX2 gene is located ∼280 kb away from the active PAN3 gene enhancer. (right) A focal genomic deletion causes an aberrant chromatin looping configuration by which an active enhancer of the PAN3 gene is retargeted (“hijacked”) and starts driving the ectopic expression of the CDX2 gene. AYA, adolescents and young adults; HR, high risk; SR, standard risk.
The authors of the 2 studies set out to define the genetic basis of a rare B-other ALL subtype characterized by a distinct gene expression profile. Kimura et al used a large collection of transcriptomic data on ∼3400 B-ALL cases across different ages and collected from different centers and identified 22 cases with this distinct transcriptional signature, constituting 0.6% of the cases in the entire cohort. Passet et al selected 302 B-other cases, mainly from 3 clinical studies (n = 723; age range:18 to 59 years), and identified 23 cases with a distinct transcriptional profile, arriving at a prevalence of 2.4% in the prospective clinical trials. By integrative WGS and WTS, the 2 studies define 2 separate underlying genetic features present in all cases with a distinct gene expression profile: (1) a focal 10-kb genomic deletion at chromosome band 17q21 causing the upstream binding transcription factor (UBTF) gene to become fused with the ataxin 7 like 3 (ATXN7L3) gene, resulting in an in-frame UBTF::ATXN7L3 gene fusion at the breakpoint junction (see figure panel B), and (2) a focal genomic deletion at 13q12 resulting in the ectopic upregulation of the caudal type homeobox 2 (CDX2) gene (see figure panel C).
The functional role of the UBTF::ATXN7L3 fusion, generated by the focal deletion at chromosome 17, has not yet been studied. Hence, it remains unknown if this fusion constitutes an oncogenic driver event or if the genomic deletion per se causes a deregulation of these 2 genes or other nearby genes. Interestingly, UBTF tandem duplications (UBTF-TD) have been previously described as recurrent genetic mutations in acute myeloid leukemia (AML), and enforced expression of the UBTF-TD in human umbilical-cord-blood cells leads to increased cellular self-renewal.7
As to the focal genomic deletion at 13q12, where FLT3 and PAN3 reside, the 2 studies show that this deletion results in ectopic expression of the CDX2 gene, located ∼30 kb downstream of FLT3. They provide evidence that the deletion at 13q12 causes an aberrant chromatin looping configuration by which an active enhancer of the PAN3 gene becomes juxtaposed to the CDX2 gene (located ∼280 kb away), thereby driving the expression of CDX2. This mechanism of aberrant gene activation is referred to as “enhancer retargeting,” or sometimes “enhancer hijacking,” and is increasingly recognized as an important mechanism underlying several disorders, including ALL.8,9 Interestingly, Yang et al10 recently described another deletion in 13q12 also removing parts of the PAN3 gene, but leaving the FLT3 promotor intact, which leads to overexpression of the FLT3 gene by the utilization of the same enhancer element within the PAN3 gene. Notably, the deletions described by Kimura et al and Passet et al remove the FLT3 promoter, hindering the expression of this gene. Thus, the 2 different types of deletions at 13q12 lead to upregulation of 2 different genes, either FLT3, a well-known player in AML and ALL pathogenesis, or CDX2. The possible oncogenic properties of CDX2 were not further investigated, but increased expression of CDX2 in patients with AML and ALL has been associated with poor prognosis and with the development of AML in mouse models.11 Recently, Yasuda et al12 also described high expression of CDX2 in B-ALL with poor risk features, present in ∼3% of their cohort (age range: 15 to 64 years), but did not study the underlying cause of the ectopic CDX2 activation.
It is presently unclear which of the 2 lesions, UBTF::ATXN7L3 or ectopic expression of CDX2, is most critical for the leukemogenic process. Based on assessment of the variant allele frequency, Kimura et al conclude that the ectopic expression of CDX2 likely is the first event. However, given their simultaneous presence in all cases, they are likely to cooperate by yet unknown mechanisms.
Are there any clinical ramifications of the 2 studies? Yes. Despite the rarity, this new B-ALL subtype (named by the authors “UBTF/CDX2 ALL” or “CDX2/UBTF ALL”) is preferentially found in AYA with a median age of 35 and 31 years, respectively.1,2 This novel subtype is associated with high-risk clinical features and a marked female preponderance, although the mechanism or mechanisms for the latter remain to be explored. From a diagnostic perspective, UBTF/CDX2 ALL (CDX2/UBTF) can be suspected based on a specific immunophenotype; most cases are negative for CD10 and CD20 and positive for cytoplasmic IgM, CD34, and CD38. However, because most, if not all, clinically relevant genetic alterations in B-ALL, including UBTF/CDX2, are detectable by combined WGS and WTS, these technologies should become the new gold standard in the diagnostic workup of B-ALL.
In conclusion, the studies by Kimura et al and Passet et al show that it is indeed worthwhile to investigate even very rare subtypes of B-ALL. They identify a new clinically relevant subtype of B-ALL and provide elegant examples of how detailed studies can reveal important insights into B-ALL pathogenesis, spurring, it is hoped, future work investigating enhancer retargeting as a mechanism underlying leukemia development. Importantly, their studies also take a crucial step toward closing the gap of the unknown genetic causes of B-other ALL.
T.F. is a cofounder, board member, and shareholder of Qlucore AB and Cantargia AB.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal