Key Points
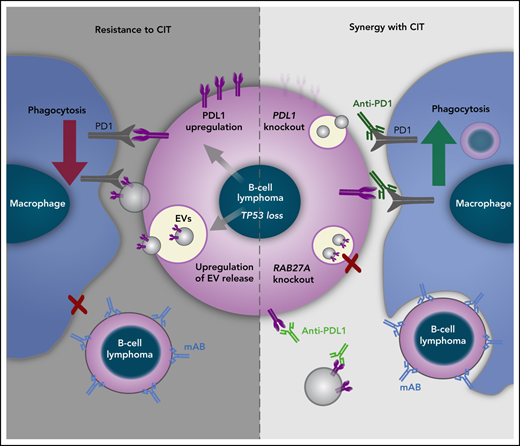
Loss of TP53 increases PD-L1 expression and EV formation from B-cell lymphoma cells.
Targeting PD-L1 and suppressing EV release overcomes TP53-mediated resistance to CIT.

Abstract
Genetic alterations in the DNA damage response (DDR) pathway are a frequent mechanism of resistance to chemoimmunotherapy (CIT) in B-cell malignancies. We have previously shown that the synergy of CIT relies on secretory crosstalk elicited by chemotherapy between the tumor cells and macrophages. Here, we show that loss of multiple different members of the DDR pathway inhibits macrophage phagocytic capacity in vitro and in vivo. Particularly, loss of TP53 led to decreased phagocytic capacity ex vivo across multiple B-cell malignancies. We demonstrate via in vivo cyclophosphamide treatment using the Eμ-TCL1 mouse model that loss of macrophage phagocytic capacity in Tp53-deleted leukemia is driven by a significant downregulation of a phagocytic transcriptomic signature using small conditional RNA sequencing. By analyzing the tumor B-cell proteome, we identified a TP53-specific upregulation of proteins associated with extracellular vesicles (EVs). We abrogated EV biogenesis in tumor B-cells via clustered regularly interspaced short palindromic repeats (CRISPR)-knockout (KO) of RAB27A and confirmed that the EVs from TP53-deleted lymphoma cells were responsible for the reduced phagocytic capacity and the in vivo CIT resistance. Furthermore, we observed that TP53 loss led to an upregulation of both PD-L1 cell surface expression and secretion of EVs by lymphoma cells. Disruption of EV bound PD-L1 by anti–PD-L1 antibodies or PD-L1 CRISPR-KO improved macrophage phagocytic capacity and in vivo therapy response. Thus, we demonstrate enhanced EV release and increased PD-L1 expression in TP53-deficient B-cell lymphomas as novel mechanisms of macrophage function alteration in CIT resistance. This study indicates the use of checkpoint inhibition in the combination treatment of B-cell malignancies with TP53 loss.
Introduction
The tumor microenvironment (TME) is characterized by multiple reciprocal interactions of malignant cells with nonmalignant stroma or immune cells.1 Particularly, macrophages are at center stage in this network, determining disease progression and therapeutic response, as well as refractory niches.2-5 In previous work, we and others could show that macrophages exert antibody-dependent cellular phagocytosis (ADCP) and represent the essential mediator of synergy in the administration of chemoimmunotherapy (CIT) of aggressive B-cell lymphoma and multiple myeloma.2,6-9 This specific combination treatment strongly increases tumor clearance by repolarization of tumor-associated macrophages from a suppressed state to an activated phenotype.2,10 CIT serves as a standard therapy in numerous B-cell malignancies and is mostly targeted at CD20, such as rituximab in combination with either fludarabine/cyclophosphamide or cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP).11,12 Despite the success of CIT in the frontline setting, therapy of relapsed or refractory disease, particularly in diffuse large B-cell lymphoma (DLBCL), still imposes a major clinical challenge. Although distinction between germinal center B-cell–like and activated B-cell subtypes and further molecular subclassification opened avenues for tailored treatment strategies, limited molecular markers are available to adapt frontline therapy to overcome primary resistant lymphoma.13,14 Immune checkpoint inhibition has revealed limited responses as monotherapy, although combination of pembrolizumab with R-CHOP in DLBCL patients was reported to show improved progression-free survival with limited toxicity.15,16
Mutations of TP53 are present in ∼20% of DLBCL and 10% of CLL, thus representing a significant subgroup of patients.13,17-19 Loss or mutation of TP53 has been identified as an important mediator of chemoresistance in a variety of malignant entities due to its central coordinating function in multiple cellular stress responses.20 Moreover, loss of TP53 mediates protumorigenic alterations in the tumor microenvironment.21,22 It remains to be clarified how alterations of the DNA damage pathway in malignant cells functionally affect the complex interactions and outcome of CIT.
Here, we primarily approached disruptions in the DNA damage response (DDR) cascade affecting the synergistic effects of CIT. Our data indicate that, particularly, TP53 functional status determines cellular crosstalk in the TME, phagocytic function, and macrophage-dependent therapeutic response to monoclonal antibodies through release of extracellular vesicles (EVs) and expression of the immune checkpoint PD-L1.
Materials and methods
Lymphoma mouse models
The hMB humanized double-hit lymphoma (DHL) and the chronic lymphocytic leukemia (CLL) Eµ-TCL1/Cd19 (Eµ-TCL1/Cd19-Tp53 wild-type (WT) (Eµ-TCL1) and Eµ-TCL1/Cd19 Cre/wt-Tp53 fl/fl (Eµ-TCL1/Tp53fl/fl) murine models were generated as described elsewhere.2,8,23-26 Detailed explanations can be found in the supplemental Material (available on the Blood Web site). Eµ-TCL1 mice were treated in vivo with cyclophosphamide (10 mg/kg) or phosphate-buffered saline (PBS) when reaching 8 to 12 weeks old and 30% of leukemic cells (CD45+CD19+CD5+ cells) in the peripheral blood.
EV isolation
EVs were extracted from supernatant of cells cultured for 24 hours at a concentration of 16.6 × 106 cells per mL in serum-free CD293 media supplemented with Glutamax 1% (85% to 100% cell viability). Supernatants were subjected to a series of centrifugation steps (1200 rpm, 5 minutes; 2900 rpm, 10 minutes; and 3500 rpm, 20 minutes) and filtered with 0.22 µm polyethersulfone (PES) membrane filters (VWR). EVs were concentrated with the Total Exosome Isolation kit (Thermo Fisher Scientific) according to the manufacturer’s instructions and ultracentrifuged (Type 45 Ti rotor, k-Factor 133, Beckman Coulter) at 100 000 g for 90 minutes at 4°C. Pellets were resuspended in cold PBS (1 µL PBS for 1 mL supernatant), dissolved by pipetting with a 1 mL syringe (Henke-Sass Wolf, Germany) with 26G 7/8-inch needle (Terumo, Germany), and immediately used for ADCPs assays or stored at −80°C. For western blot analysis, EV pellet was directly lysed.
Results
Macrophage phagocytic capacity upon chemotherapy is diminished with dependence of DDR pathway mediators in the leukemic compartment
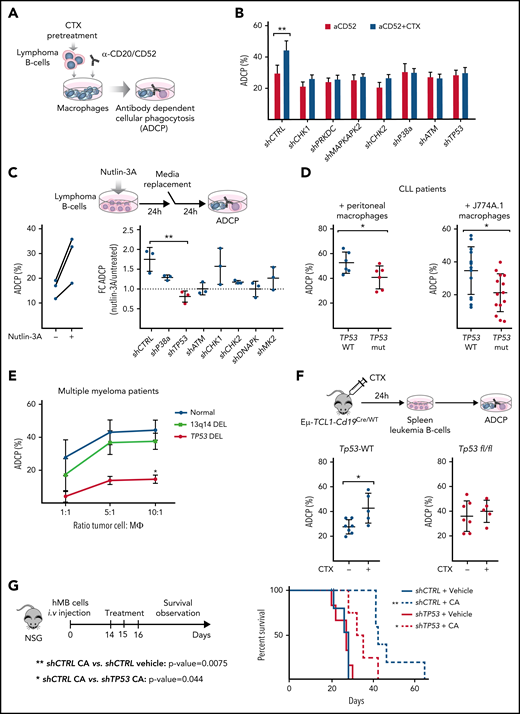
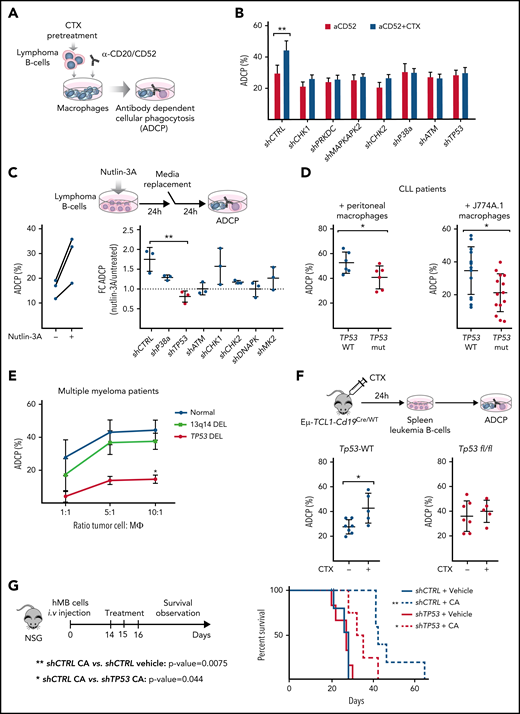
To dissect the mechanism of the synergistic interaction between alkylating chemotherapy and monoclonal antibodies,2,6,24 we disrupted key components of the DDR pathway in 2 different aggressive B-cell lymphoma models: the hMB humanized DHL model24 and the Myd88 p.L252P-driven DLBCL model.27 In both cases, we generated short hairpin RNA (shRNA)-mediated knockdowns of genes that code for the key DDR mediators: checkpoint kinase 1 (CHK1), CHK2, DNA-dependent protein kinase (PRKDC), MAPK-activated protein kinase 2 (MAPKAPK2), p38-α, ATM, and TP53 (supplemental Figure 1A-B). We exposed shRNA-defined DDR component-deficient cells to mafosfamide, an in vitro alkylating agent, as surrogate for the effect of cyclophosphamide. We then determined the ADCP rate using alemtuzumab or 18B12 anti-CD20 antibody in coculture assays with various sources of macrophages (Figure 1A). We could observe that chemotherapy pretreatment of hMB lymphoma B-cells significantly enhanced their susceptibility for phagocytic engulfment as compared with untreated shRNA-CTRL (shCTRL)-hMB cells (Figure 1B). However, disruption of the DDR led to an abrogation of increased phagocytosis compared with chemotherapy-treated shCTRL-hMB cells (Figure 1B). To employ an independent model of aggressive lymphoma, we used the Myd88 p.L252P-driven DLBCL model, where we confirmed that combination treatment of mafosfamide with 18B12 improved the phagocytosis of shCTRL Myd88 p.L252P cells by primary peritoneal and bone marrow–derived macrophages, as well as by the macrophage cell line J774A.1 (supplemental Figure 1C). In addition, Chk1-, Prkdc-, and MAPKAPK2-deficient lymphoma cells were sensitized to mafosfamide treatment (supplemental Figure 1D). However, Tp53-deficient cells displayed the highest resistance toward CIT (supplemental Figure 1D).
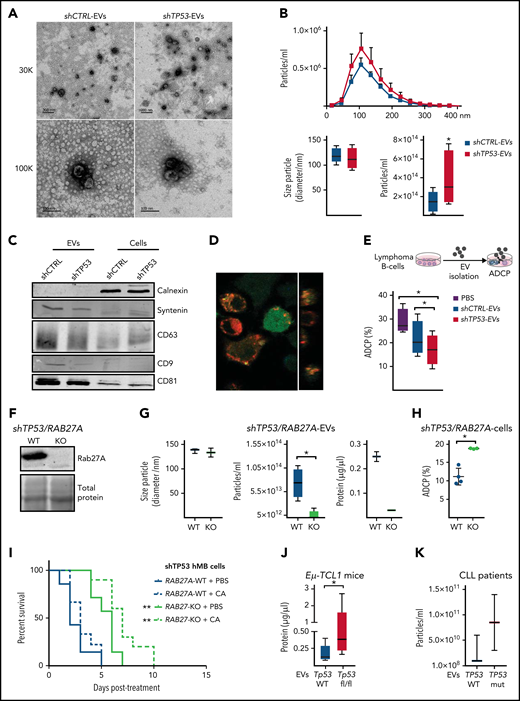
TP53 loss in lymphoma cells impairs macrophage ability to phagocytose tumor B cells. (A) Schematic representation of an ADCP assay. (B) Alemtuzumab (anti-CD52)-mediated ADCP of empty vector (shCTRL) and the indicated DDR-knockdown hMB cells, pretreated or not with 3 µM mafosfamide (CTX) and cocultured with peritoneal macrophages (3 independent experiments, 5 replicates per experiment). (C) ADCP percent of shCTRL hMB cells, pretreated or not with nutlin-3A (left panel) (n = 3). Right panel shows ADCP fold change (FC) induced upon nutlin-3A pretreatment on the indicated DDR-knockdown hMB cells (n = 3). (D) ADCP assay of CLL cells from patients with TP53 WT and mutant expression (TP53 mut) on the presence of peritoneal macrophages and alemtuzumab (left) or mCherry+ J77A4.1 macrophages and 10 mg rituximab and 3 µM mafosfamide (n = 6) or J77A4.1 macrophages (n = 15). (E) Daratumumab (anti-CD38 antibody)-mediated ADCP of multiple myeloma (MM) tumor cells obtained from patients with normal TP53 expression (normal karyotype or 13q14 deletion) and from patients with TP53 deletion. Tumor cells were cocultured with autologous macrophages in the indicated ratios (10:1 ratio, del13q14DEL vs TP53DEL P = .025; n = 3). (F) Eµ-TCL1/wt and Eµ-TCL1/Tp53fl/fl leukemic mice were treated or not with 10 mg/kg cyclophosphamide (CTX) for 24 hours. Then, the spleen leukemic cells were isolated and used to perform an ADCP in the presence of peritoneal macrophages (n = 5-7). (G) Kaplan-Meier analysis comparing the survival of shCTRL and shTP53 hMB–transplanted nonobese diabetes scid gamma mice receiving cyclophosphamide and alemtuzumab (CA) as CIT combination. PBS was used as control. The treatment was given IP 10 days after IV hMB cell injection (n = 5-6). (*P < .05, **P ≤ .01). IP, intraperitoneal.
TP53 loss in lymphoma cells impairs macrophage ability to phagocytose tumor B cells. (A) Schematic representation of an ADCP assay. (B) Alemtuzumab (anti-CD52)-mediated ADCP of empty vector (shCTRL) and the indicated DDR-knockdown hMB cells, pretreated or not with 3 µM mafosfamide (CTX) and cocultured with peritoneal macrophages (3 independent experiments, 5 replicates per experiment). (C) ADCP percent of shCTRL hMB cells, pretreated or not with nutlin-3A (left panel) (n = 3). Right panel shows ADCP fold change (FC) induced upon nutlin-3A pretreatment on the indicated DDR-knockdown hMB cells (n = 3). (D) ADCP assay of CLL cells from patients with TP53 WT and mutant expression (TP53 mut) on the presence of peritoneal macrophages and alemtuzumab (left) or mCherry+ J77A4.1 macrophages and 10 mg rituximab and 3 µM mafosfamide (n = 6) or J77A4.1 macrophages (n = 15). (E) Daratumumab (anti-CD38 antibody)-mediated ADCP of multiple myeloma (MM) tumor cells obtained from patients with normal TP53 expression (normal karyotype or 13q14 deletion) and from patients with TP53 deletion. Tumor cells were cocultured with autologous macrophages in the indicated ratios (10:1 ratio, del13q14DEL vs TP53DEL P = .025; n = 3). (F) Eµ-TCL1/wt and Eµ-TCL1/Tp53fl/fl leukemic mice were treated or not with 10 mg/kg cyclophosphamide (CTX) for 24 hours. Then, the spleen leukemic cells were isolated and used to perform an ADCP in the presence of peritoneal macrophages (n = 5-7). (G) Kaplan-Meier analysis comparing the survival of shCTRL and shTP53 hMB–transplanted nonobese diabetes scid gamma mice receiving cyclophosphamide and alemtuzumab (CA) as CIT combination. PBS was used as control. The treatment was given IP 10 days after IV hMB cell injection (n = 5-6). (*P < .05, **P ≤ .01). IP, intraperitoneal.
We speculated that the disruption of CIT synergy response when altering DDR components could be a result of insufficient p53 activation. Thus, we used nutlin-3A as an indirect activator of p53 and observed that nutlin-3A similarly modified the ADCP of control, CHK1-, CHK2-, p38a-, and MAPKAPK2-deficient hMB cells but left TP53-deficient lymphoma cells unaffected, which exhibited a significantly reduced phagocytosis level (Figure 1C). Furthermore, nutlin-3A treatment of PRKDC- and ATM-deficient cells did not alter macrophage phagocytic capacity, which correlates with the absence of p53 induction by nutlin-3A (supplemental Figure 1E-F). Altogether, these results indicate that p53-axis functions as a key node in lymphoma cells to influence the response of macrophages to CIT.
To further confirm our observation, we used primary CLL patient samples defined by their TP53 status (Figure 1D; supplemental Figure 1E; supplemental Table 1.3). Notably, macrophages cocultured in the presence of monoclonal antibodies alemtuzumab or rituximab with TP53-mutant CLL cells showed significantly lower ADCP than macrophages cocultured with TP53-WT cells, irrespective of TP53 mutant clone size (Figure 1D; supplemental Figure 1H). Likewise, the effect of TP53 on tumor cell–macrophage crosstalk was investigated using primary malignant MM patient cells cocultured with autologous macrophages in the presence of daratumumab (Figure 1E).28 Macrophages were able to phagocytose ∼50% of MM cells from patients with normal karyotype and del13q14-aberration but displayed a significantly reduced phagocytosis of TP53-deficient MM cells (Figure 1E) that was particularly lower in the higher ratios (10:1 ratio, del13q14DEL vs TP53DEL, P = .025).29 The relevance of TP53 as a central regulator of microenvironment-dependent treatment response in vivo was validated by utilizing the Eµ-TCL1 mouse model of CLL. Here, leukemic Eµ-TCL1/Cd19-Tp53 WT (Eµ-TCL1) and Eµ-TCL1/Cd19Cre/wt-Tp53 fl/fl (Eµ-TCL1/Tp53fl/fl) were treated with cyclophosphamide. After 24 hours, leukemic cells were obtained for ex vivo ADCP assessment targeted by the antimurine CD20 antibody. In line with our previous observations, in vivo chemotherapy treatment significantly improved the ADCP of Eµ-TCL1–derived leukemia cells, whereas no phagocytic improvement was found in Eµ-TCL1/Tp53fl/fl–derived cells (Figure 1F). These results indicate that loss of TP53 switches malignant B-cells toward a refractory state against CIT. To verify our hypothesis, we studied the in vivo response to CIT with the humanized mouse model of DHL. Specifically, shTP53-transduced hMB alongside shCTRL cells were transplanted into immunodeficient NSG recipient mice and CIT treatment (CA) was initiated upon disease onset (Figure 1G). CIT resulted in a significantly increased survival of mice transplanted with shCTRL-hMB cells (CA median, 41.5 days vs PBS median, 28 days; P = .0075). However, the response of mice bearing the shTP53-hMB cells to the combination therapy showed a significantly shorter overall survival (CA median, 33.5 days; P = .044) compared with shCTRL-hMB cells (Figure 1G). In summary, TP53 expression serves as a central regulatory node for interactions with macrophages in the TME, particularly in determining response to CIT in B-cell malignancies.
Tp53 dependent interactions within the lymphoma TME and its effects on macrophage phenotype and function
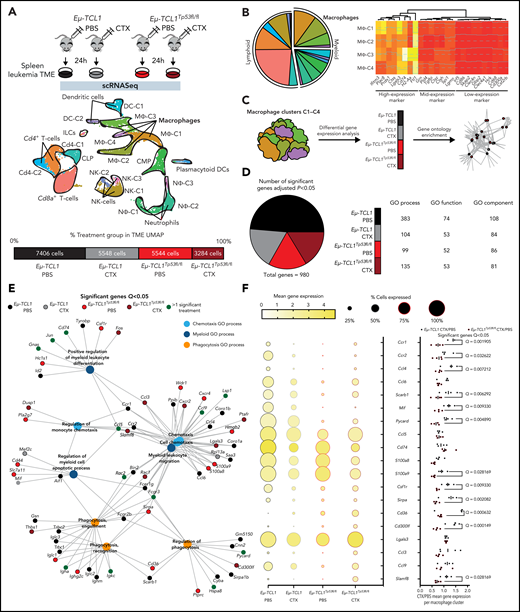
To identify potential p53-dependent pathway alterations induced between malignant B-cells and the TME, we leveraged CD19+ cell–depleted TME cells from both Eµ-TCL1 and T Eµ-TCL1/Tp53fl/fl models under in vivo chemotherapeutic treatment and subjected them to single-cell RNA sequencing (scRNA-seq) analysis. We obtained 21 782 cells across conditions (Figure 2A; supplemental Figure 2A), with 4 distinct macrophage clusters, which were comprised of the largest TME compartment (Figure 2B, left; supplemental Figure 2B) and sharing a largely similar expression of macrophage marker genes across the 4 clusters (Figure 2B, right; supplemental Figure 2C). We therefore decided to combine data from all macrophage clusters together followed by GO enrichment analysis stratified by treatment and genotype to identify the key pathways regulating macrophage function. (Figure 2C). Here, we identified that of the 980 significantly regulated genes in the dataset, 477 were Eµ-TCL1 PBS, 178 Eµ-TCL1 CTX, 165 Eµ-TCL1/Tp53fl/fl PBS, and 160 Eµ-TCL1/Tp53fl/fl CTX (Figure 2D, left) and that the GO terms across process/function/component were all largely dominated by Eµ-TCL1 PBS (Figure 2D, right). However, we observed an enrichment for chemotaxis/phagocytic/myeloid GO process terms that were driven by overexpression under Eµ-TCL1 PBS and Eµ-TCL1/Tp53fl/fl CTX treatment (Figure 2E). Under closer inspection, we observed both reduced expression and percentage of positive cells in Eµ-TCL1 PBS and Eµ-TCL1CTX treatment groups, whereas we observed the opposite under Eµ-TCL1/Tp53fl/fl PBS and Eµ-TCL1/Tp53fl/fl CTX treatment (Figure 2F, middle). Furthermore, after CTX/PBS gene expression calculation for the original 4 macrophage clusters in Eµ-TCL1 and Eµ-TCL1/Tp53fl/fl, respectively, we further observed in the context of Tp53 loss that these chemotaxis/phagocytic/myeloid genes are significantly downregulated in relation to Eµ-TCL1 TME cells, such as Ccr1/2 (Q, 0.001905; Q, 0.032622), Mif/Pycard (Q, 0.009330; Q, 0.004890), and Csfr1/Cd36 (Q, 0.009330; Q, 0.000632) (Figure 2F, right). In all, this scRNA-seq analysis suggests that loss of Tp53 on malignant B-cells under in vivo chemotherapy treatment drives fundamental changes to the macrophage TME, namely loss of phagocytic function and downregulation of chemotaxis.
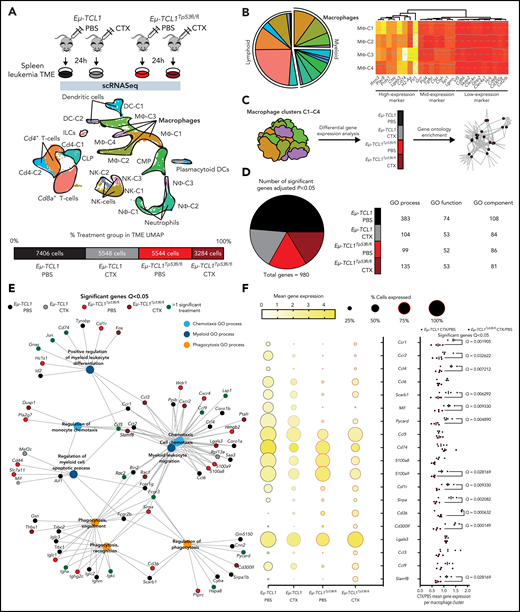
Characterization of TME-macrophage functions upon chemotherapy on Eμ-TCL1 mice.Eμ-TCL1-Cd19Cre-TP53wt (Eμ-TCL1) and Eμ-TCL1-Cd19Cre-TP53fl/fl (Eμ-TCL1) leukemic mice (Cd45+Cd19+Cd5+ cells on blood >30%) were IP injected 10 mg/kg cyclophosphamide (CTX) or PBS for 24 hours. (A) Schematic representation of the experimental design (upper panel). Splenocytes were isolated from the spleens of each treatment group, followed by MACS B-cell depletion (Cd19− cells) before scRNA-seq. Integrated Uniform Manifold Approximation and Projection (UMAP) dimension reduction plot of all treatment groups (n = 21 782 cells) (center panel). Cells are colored by the clusters determined by cell type. Percentage and total cell number of each treatment group contributing to the integrated UMAP (lower panel). (B) Pie chart representing the proportion of each cluster across all genotypes and treatment groups (left panel). Heat map showing the cell marker gene expression profiles of all macrophage clusters (MΦ-1, MΦ-2, MΦ-3, and MΦ-4) across all genotypes and treatment groups (right panel). (C) Diagram representing the gene ontology (GO) term analysis strategy across both genotype and treatment groups. (D) Pie chart showing the percentage of genotype and treatment group significantly regulated genes (left panel). GO process/function/component table showing the number of significant GO terms per genotype and treatment group (right panel). (E) GGnet plot of chemotaxsis/myeloid/phagocytosis GO process terms enriched in the macrophage clusters together with notation of the genotype and treatment group of significant genes of the GO terms (black, Eµ-TCL1 PBS; gray, Eµ-TCL1 CTX; light red, Eµ-TCL1Tp53fl/fl; dark red, Eµ-TCL1Tp53fl/fl CTX; dark green, >1 significant group). (F) Ball plot showing the percentage of positive cells and average expression across all macrophage cluster cells per genotype and treatment group for key GO process genes (left panel). Bar graph showing the fold change treatment normalized (CTX/PBS) per genotype per macrophage cluster (MΦ-1, MΦ-2, MΦ-3, and MΦ-4) for the same genes as in the left panel (right panel). Significant differences calculated either in Seurat (B/D), GO Gorilla (D/E), or GraphPad (F), respectively. In all instances, adjusted P values from the respective methods were used (see “Materials and methods”). IP, intraperitoneal; MACS, magnetic-activated cell sorting.
Characterization of TME-macrophage functions upon chemotherapy on Eμ-TCL1 mice.Eμ-TCL1-Cd19Cre-TP53wt (Eμ-TCL1) and Eμ-TCL1-Cd19Cre-TP53fl/fl (Eμ-TCL1) leukemic mice (Cd45+Cd19+Cd5+ cells on blood >30%) were IP injected 10 mg/kg cyclophosphamide (CTX) or PBS for 24 hours. (A) Schematic representation of the experimental design (upper panel). Splenocytes were isolated from the spleens of each treatment group, followed by MACS B-cell depletion (Cd19− cells) before scRNA-seq. Integrated Uniform Manifold Approximation and Projection (UMAP) dimension reduction plot of all treatment groups (n = 21 782 cells) (center panel). Cells are colored by the clusters determined by cell type. Percentage and total cell number of each treatment group contributing to the integrated UMAP (lower panel). (B) Pie chart representing the proportion of each cluster across all genotypes and treatment groups (left panel). Heat map showing the cell marker gene expression profiles of all macrophage clusters (MΦ-1, MΦ-2, MΦ-3, and MΦ-4) across all genotypes and treatment groups (right panel). (C) Diagram representing the gene ontology (GO) term analysis strategy across both genotype and treatment groups. (D) Pie chart showing the percentage of genotype and treatment group significantly regulated genes (left panel). GO process/function/component table showing the number of significant GO terms per genotype and treatment group (right panel). (E) GGnet plot of chemotaxsis/myeloid/phagocytosis GO process terms enriched in the macrophage clusters together with notation of the genotype and treatment group of significant genes of the GO terms (black, Eµ-TCL1 PBS; gray, Eµ-TCL1 CTX; light red, Eµ-TCL1Tp53fl/fl; dark red, Eµ-TCL1Tp53fl/fl CTX; dark green, >1 significant group). (F) Ball plot showing the percentage of positive cells and average expression across all macrophage cluster cells per genotype and treatment group for key GO process genes (left panel). Bar graph showing the fold change treatment normalized (CTX/PBS) per genotype per macrophage cluster (MΦ-1, MΦ-2, MΦ-3, and MΦ-4) for the same genes as in the left panel (right panel). Significant differences calculated either in Seurat (B/D), GO Gorilla (D/E), or GraphPad (F), respectively. In all instances, adjusted P values from the respective methods were used (see “Materials and methods”). IP, intraperitoneal; MACS, magnetic-activated cell sorting.
Chemotherapy treatment modifies the proteomic profile of lymphoma B-cells in a p53-dependent manner and indicates altered EV formation
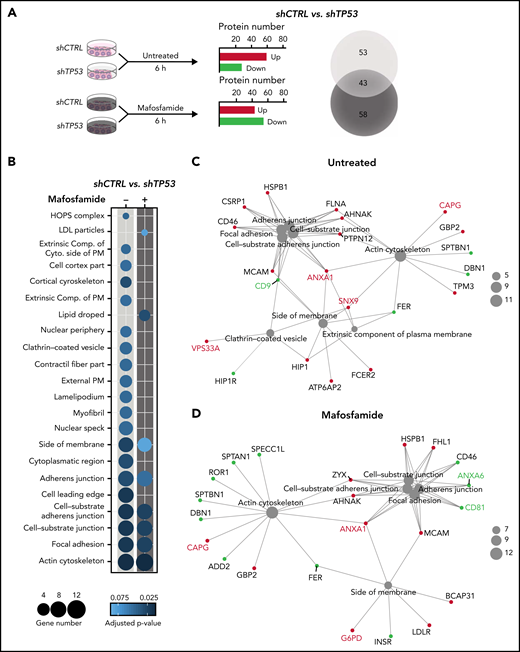
We identified TP53 not to specifically alter cytokine mediated activations of macrophages (supplemental Figure 3A); however, significant effects by conditioned media of TP53-deficient lymphomas indicated a functional role of soluble crosstalk (supplemental Figure 3B). To identify the mechanism of this specific interaction, we assessed shCTRL and shTP53 tumor B-cells’ proteomic landscape after mafosfamide treatment (1 µM, 6 hours) by a mass spectrometry label-free protein quantification approach (Figure 3A). In total, >4000 proteins were detected in every condition (supplemental Table 3.1). Hierarchical agglomerative clustering clearly differentiated the shCTRL from the shTP53 proteome (supplemental Figure 3A). Our results showed that TP53 depletion profoundly influenced the proteomic profile of lymphoma B-cells, showing a downregulation of 36 proteins and the upregulation of another 60, compared with the control cells (Figure 3A). Interestingly, we observed that mafosfamide treatment differently modified the proteomic profile of TP53-deficient and control cells (supplemental Figure 3B) and produced a shift in the number of up- and downregulated proteins controlled by TP53 (Figure 3A; supplemental Figure 3D-F). Analyzing GO terms of significantly (P-adjust ≤0.1) regulated proteins (supplemental Table 3.2), we observed that loss of TP53 expression mainly correlated to changes on actin cytoskeleton, extracellular plasma membrane, and vesicle-related terms, composed of proteins such as CD9, vacuolar protein sorting 33A (VPS33A), and sorting nexin 9 (SNX9) (Figure 3B-C). Similarly, mafosfamide treatment of shTP53 cells differently regulated proteins associated to actin cytoskeleton and cell adherens junction processes, displaying altered expression levels of EV-associated proteins, such as annexins 1 and 6 (ANXA1 and ANXA6) and the tetraspanin CD81 (Figure 3B-D). Therefore, proteomic analysis indicated that TP53 loss in lymphoma B-cells involves actin cytoskeleton reorganization and alterations in the formation of EVs, which we hypothesize could be responsible for the communication between the malignant B-cells and the TME macrophages.
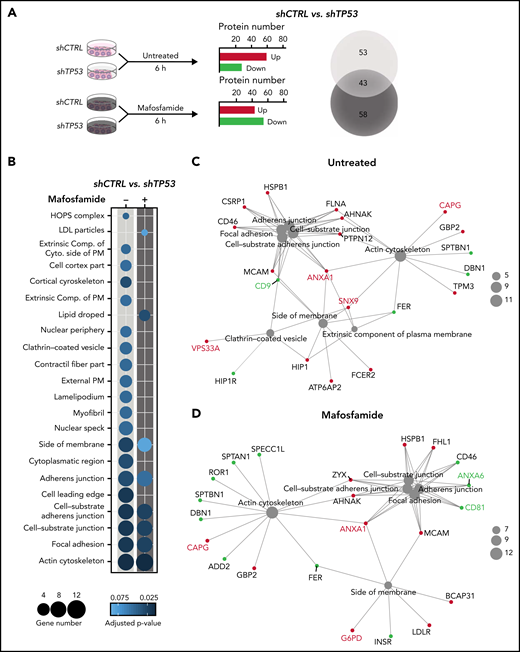
Chemotherapy-rewiring of control and TP53-deficient lymphoma B-cells’ proteome. (A-D) shCTRL and shTP53 hMB cells were treated with 3 µM mafosfamide or vehicle (untreated) for 6 hours, and their proteomic profiles were analyzed by mass spectrometry label-free protein quantification (n = 3). (A) Workflow of the experiment (left panel). Bar plot of the number of up- and downregulated proteins that are differentially expressed in shTP53 hMB cells in comparison with shCTRL hMB cells, either in untreated or mafosfamide-treated condition (central panel). Venn diagram illustrating the total number of significant proteins specifically or commonly regulated by TP53 in either untreated or mafosfamide-treated condition. (B-D) Enriched cellular component GO terms identified in the analysis of significantly regulated proteins by TP53 expression in untreated (-) and mafosfamdie-treated (+) hMB cells. (B) Sphere sizes represent the number of proteins associated to every GO term, whereas the color indicates the adjusted P-value (lower panel). (C-D) Network interaction depicting the linkage of proteins and selected CC-GO terms identified in the comparison analysis between TP53-deficient and control hMB cells upon (C) vehicle (untreated) or (D) mafosfamide treatment. Sphere sizes represent the number of proteins associated to every GO term. Green and red nodes highlight down- and upregulated proteins, respectively. Proteins involved in EV biogenesis or release have been highlighted.
Chemotherapy-rewiring of control and TP53-deficient lymphoma B-cells’ proteome. (A-D) shCTRL and shTP53 hMB cells were treated with 3 µM mafosfamide or vehicle (untreated) for 6 hours, and their proteomic profiles were analyzed by mass spectrometry label-free protein quantification (n = 3). (A) Workflow of the experiment (left panel). Bar plot of the number of up- and downregulated proteins that are differentially expressed in shTP53 hMB cells in comparison with shCTRL hMB cells, either in untreated or mafosfamide-treated condition (central panel). Venn diagram illustrating the total number of significant proteins specifically or commonly regulated by TP53 in either untreated or mafosfamide-treated condition. (B-D) Enriched cellular component GO terms identified in the analysis of significantly regulated proteins by TP53 expression in untreated (-) and mafosfamdie-treated (+) hMB cells. (B) Sphere sizes represent the number of proteins associated to every GO term, whereas the color indicates the adjusted P-value (lower panel). (C-D) Network interaction depicting the linkage of proteins and selected CC-GO terms identified in the comparison analysis between TP53-deficient and control hMB cells upon (C) vehicle (untreated) or (D) mafosfamide treatment. Sphere sizes represent the number of proteins associated to every GO term. Green and red nodes highlight down- and upregulated proteins, respectively. Proteins involved in EV biogenesis or release have been highlighted.
Secretion of EVs from TP53-deficient lymphoma B-cells is increased and reduces the antitumor effector function of macrophages
EVs have emerged as an important mechanism of cellular communication between tumor and stroma cells.30-32 To assess the relevance of the proteomic findings, we sought to characterize EVs produced by control and TP53-deficient lymphoma B-cells. EVs isolated from lymphoma B-cell supernatants showed typical cup-shaped morphology with a size ranging from 50 to 200 nm (Figure 4A). Loss of TP53 did not alter vesicle size (Figure 4B) or induce significant changes in microRNA (miRNA) abundance (supplemental Table 4.1). However, NTA showed that TP53-deficient cells released significantly higher numbers of EVs than the control cells (Figure 4B). We characterized EV-protein composition by immunoblot, detecting an enriched expression of the EV markers CD81, CD9, CD63, and syntenin compared with the respective cell source, whereas calnexin, a negative marker for EVs, was only present on the cell lysate samples (Figure 4C).30 To determine whether TP53-deficient cell-derived EVs could affect macrophage properties, we exposed different types of macrophages to EVs isolated from shCTRL (shCTRL-EVs) or shTP53 lymphoma B-cells (shTP53-EVs). Our results showed that shTP53-EVs were engulfed by macrophages in a similar way as the shCTRL-EVs (Figure 4D; supplemental Figure 4A). However, macrophages exposed to shTP53-EVs during the coculture exhibited a significantly reduced phagocytic capacity of lymphoma B-cells, whereas they were not significantly affected by shCTRL-EVs (Figure 4E). TP53-dependent inhibition on the ADCP persisted even when the concentration of shTP53-EVs added in the coculture was reduced by 90% (supplemental Figure 4B). In parallel, we excluded EV-mediated effects on lymphoma B-cell viability (supplemental Figure 4C). To clarify if shTP53-EVs were affecting general macrophage phagocytic capacity, we performed a bead-based phagocytosis assay, where macrophages were exposed to shCTRL- or shTP53-EVs 16 hours prior to adding DyLight680-labeled latex beads (supplemental Figure 4D). Bead phagocytosis was unaffected by the presence of shTP53-EVs (supplemental Figure 4D). Thus, shTP53-EVs specifically abrogated Fc receptor– dependent macrophage antitumor effects, such as ADCP. Next, to address the functional role of EVs on the lymphoma B-cell-macrophage crosstalk, we generated lymphoma B-cells unable to release EVs by depleting RAB27A expression via the CRISPR/Cas9 approach (Figure 4F).33 We proved that RAB27A knockout cells (RAB27A-KO) showed a significantly reduced ability to produce EVs (Figure 4G).
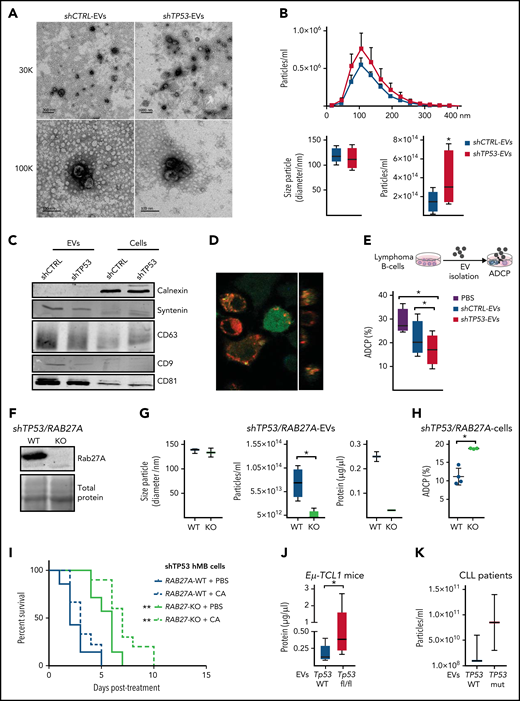
Features and functions of EVs derived from TP53-deficient and control lymphoma B cells. (A-G) EVs were isolated from TP53-deficient (shTP53-EVs) and control (shCTRL-EVs) hMB lymphoma B-cells. (A) Representative TEM images. Thirty thousand and 100 000 magnifications are shown. (B) Nanoparticle tracking analysis (NTA) of the EVs distribution of the particles according to their size and concentration (upper panel). Box plots showing size particle and particle concentration of the isolated EVs (lower panel; n = 6). (C) Immunoblot analysis of the isolated EVs and their corresponding cell lysates using the indicated antibodies. (D) Confocal image showing DiD-labeled EV (red) uptake by GFP+J774A.1 macrophages (green) after 16 hours in culture. Orthogonal view of the same image confirms the intracellular presence of the EVs (right panel). (E) Alemtuzumab-mediated ADCP of control hMB cells cocultured with J774A.1 macrophages and shCTRL-EVs, shTP53-EVs, or vehicle (PBS) (n = 4). (F-I) RAB27A expression was depleted by clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) approached in shTP53 hMB cells. (F) Immunoblotting detection of Rab27a protein in shTP53/RAB27A-WT and -KO hMB cells. The corresponding total protein staining of the samples as a protein loading control is shown. (G) Box plots of NTA determinations (left and middle panel) and protein concentration (right panel) of EVs obtained from shTP53/RAB27A-WT and -KO EVs (n = 3). (H) Alemtuzumab-mediated ADCP of different clones of both, shTP53/RAB27A-WT (n = 4) and –KO (n = 3) hMB cells cocultured with J774A.1 macrophages. All the graphics showed the mean plus or minus SD of at least 3 independent experiments. (I) Kaplan-Meier analysis comparing the survival of shTP53/RAB27A-WT and -KO hMB transplanted NSG mice receiving cyclophosphamide and alemtuzumab (CA) as CIT combination. PBS was used as control. The treatment was given IP 10 days after IV hMB cell injection (n = 7-10). (J) Protein determination of EVs derived from CD19+cells isolated from the spleen of sick Eµ-TCL1 WT and Eµ-TCL1/Tp53fl/fl mice (n = 5-8). (K) Concentration of EVs isolated from primary CLL patient cells (n = 2-3) (*P < .05, **P ≤ .01). IP, intraperitoneal; SD, standard deviation.
Features and functions of EVs derived from TP53-deficient and control lymphoma B cells. (A-G) EVs were isolated from TP53-deficient (shTP53-EVs) and control (shCTRL-EVs) hMB lymphoma B-cells. (A) Representative TEM images. Thirty thousand and 100 000 magnifications are shown. (B) Nanoparticle tracking analysis (NTA) of the EVs distribution of the particles according to their size and concentration (upper panel). Box plots showing size particle and particle concentration of the isolated EVs (lower panel; n = 6). (C) Immunoblot analysis of the isolated EVs and their corresponding cell lysates using the indicated antibodies. (D) Confocal image showing DiD-labeled EV (red) uptake by GFP+J774A.1 macrophages (green) after 16 hours in culture. Orthogonal view of the same image confirms the intracellular presence of the EVs (right panel). (E) Alemtuzumab-mediated ADCP of control hMB cells cocultured with J774A.1 macrophages and shCTRL-EVs, shTP53-EVs, or vehicle (PBS) (n = 4). (F-I) RAB27A expression was depleted by clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) approached in shTP53 hMB cells. (F) Immunoblotting detection of Rab27a protein in shTP53/RAB27A-WT and -KO hMB cells. The corresponding total protein staining of the samples as a protein loading control is shown. (G) Box plots of NTA determinations (left and middle panel) and protein concentration (right panel) of EVs obtained from shTP53/RAB27A-WT and -KO EVs (n = 3). (H) Alemtuzumab-mediated ADCP of different clones of both, shTP53/RAB27A-WT (n = 4) and –KO (n = 3) hMB cells cocultured with J774A.1 macrophages. All the graphics showed the mean plus or minus SD of at least 3 independent experiments. (I) Kaplan-Meier analysis comparing the survival of shTP53/RAB27A-WT and -KO hMB transplanted NSG mice receiving cyclophosphamide and alemtuzumab (CA) as CIT combination. PBS was used as control. The treatment was given IP 10 days after IV hMB cell injection (n = 7-10). (J) Protein determination of EVs derived from CD19+cells isolated from the spleen of sick Eµ-TCL1 WT and Eµ-TCL1/Tp53fl/fl mice (n = 5-8). (K) Concentration of EVs isolated from primary CLL patient cells (n = 2-3) (*P < .05, **P ≤ .01). IP, intraperitoneal; SD, standard deviation.
Next, we proved that ADCP targeting shTP53/RAB27A-KO tumor cells was significantly higher than targeting shTP53/WT controls (Figure 4H). To validate our EV-related results in vivo, we used the hMB humanized mouse model of DHL, transplanting mice with shTP53/RAB27A-KO or shTP53/RAB27A-WT hMB cells and using CIT or PBS (control) as treatment. We observed that mice transplanted with lymphoma B-cells unable to release EVs displayed significantly longer overall survival after CIT (CA+ shTP53/RAB27A-KO median equals 21 plus or minus 0.48 days vs CA+ shTP53/RAB27A-WT median equals 17 plus or minus 0.47 days; P ≤ .0001) (Figure 4I). To further support our hypothesis, we also confirmed a significant increase in total number of EVs produced by TP53-deficient primary murine CLL (Eµ-TCL1) cells (Figure 4J) and an upward trend in TP53-mutant primary CLL cells vs WT cells (Figure 4K). In conclusion, we show that EV formation is enhanced by loss of TP53 and leads to suppression of macrophage effector function toward malignant cells.
Checkpoint pathway inhibition via the PD1/PD-L1 signaling axis restores the CIT-induced macrophage phagocytosis of TP53-deficient lymphoma B-cells
Given the diminished phagocytic capacity of macrophages in the presence of EVs produced by TP53-deficient malignant B-cells, we considered the possibility that the EVs secreted by shCTRL and shTP53 cells displayed different expression levels of “dońt eat me” and related immune checkpoint molecules. Our data confirmed the absence of CD47 and the low expression of CD200, respectively, but the presence of PD-L1 on leukemic B cells-EVs (Figure 5A). Next, by using the anti–PD-L1 antibody atezolizumab, PD-L1 was blocked on the EVs before their addition to the coculture of macrophages and leukemic B-cells (Figure 5B). Remarkably, neutralizing PD-L1 on the shTP53-EVs, we could restore macrophage antitumor capacity assessed by ADCP. This observation was confirmed by using EVs obtained from PD-L1–knockout (PD-L1–KO) hMB cells, which do not impair macrophage antitumor function (Figure 5C-D). Thus, PD-L1 expression on shTP53-EVs mediate suppression of macrophage phagocytic activity through PD1 expressed on macrophages (supplemental Figure 5A-B).
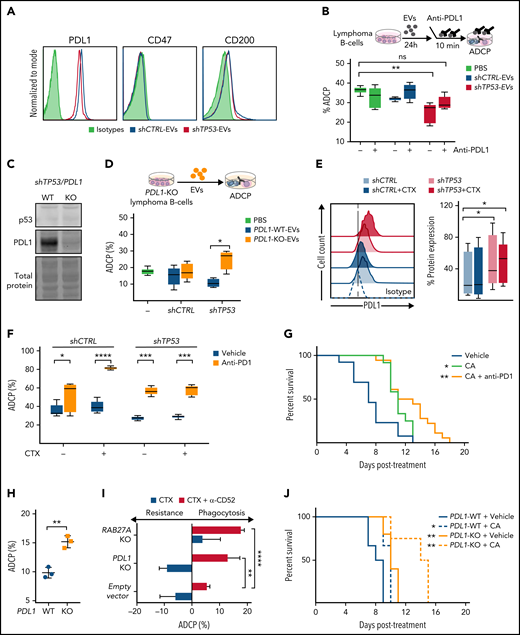
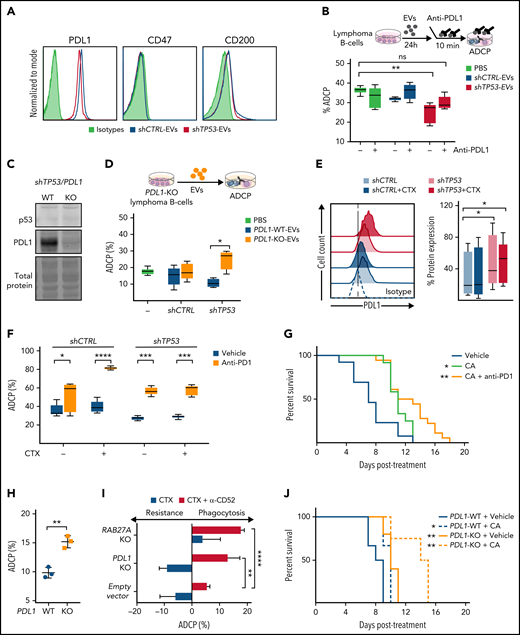
Identification of EVs and PD-L1 expression roles on the resistance to chemotherapy in TP53-deficeint B-cell lymphomas. (A) Flow cytometer analysis of shCTRL− and shTP53-EVs bound to polystyrene microspheres and stained for the indicated proteins or the correspondent isotype control (1 representative experiment of 3). (B) Alemtuzumab-mediated ADCP assay with normal hMB cells and J774A.1 macrophages in the presence of EVs derived from shCTRL and shTP53 hMB cells that were incubated (10 minutes) with anti–PD-L1 antibody (atezolizumab) prior to their addition to the coculture. The same concentration of anti–PD-L1 antibody diluted in PBS was used as control. Data shows 1 representative experiment of 3, with 5 replicates. (C) Immunoblotting detection of p53 and PD-L1 protein in shTP53/PD-L1–WT and -KO hMB cells. The corresponding total protein staining of the samples as a protein-loading control is shown. (D) Alemtuzumab-mediated ADCP assay with normal hMB cells with J774A.1 macrophages in the presence of shTP53/PD-L1–WT and -KO EVs; PBS was used as control. Data shows 1 representative experiment of 3, with 5 replicates. (E) Flow cell cytometer detection of PD-L1 in shCTRL and shTP53 hMB cells, treated with mafosfamide (CTX) or vehicle for 24 hours. Left panel shows half offset histogram representation of 1 representative experiment. Right panel displays PD-L1 protein expression percentage from 6 independent experiments. (F) Anti-CD20 (18B12)-mediated ADCP of shCTRL and shTP53 hMB cells pretreated or not with mafosfamide and cocultured with peritoneal macrophages exposed to anti-PD1 antibody (GS-696882) from 4 hours prior to the assay. Data shows 1 representative experiment of 3, with 5 replicates. (G) Survival curve of NSG mice IV injected with shTP53 hMB tumor cells treated IP with cyclophosphamide and alemtuzumab (CA) or CA and anti-PD1 antibody (GS-696882). PBS was used as control treatment (n = 12-18). (H) Alemtuzumab-mediated ADCP of different shTP53/PD-L1–WT and -KO clones coculture with J774A.1 macrophages (n = 3). (I) Number of mafosfamide (CTX) pretreated GFP+ hMB cells (shTP53/PD-L1–KO, shTP53/RAB27A-KO, and the corresponding shTP53 empty vector) remaining in the coculture with J774A.1 macrophages after ADCP assay performed in combination or not with alemtuzumab and normalized to the amount of hMB cells in a hMB single culture. Data shows 1 representative experiment, with 5 replicates. (J) Survival curve of NSG mice IV injected with shTP53/PD-L1-WT or shTP53/PD-L1-KO hMB tumor cells and IP treated after 10 days with cyclophosphamide and alemtuzumab (CA) or PBS as control treatment (n = 3-6). *P < .05, **P ≤ .01, ***P ≤ .001, ****P ≤ .0001. IP, intraperitoneal.
Identification of EVs and PD-L1 expression roles on the resistance to chemotherapy in TP53-deficeint B-cell lymphomas. (A) Flow cytometer analysis of shCTRL− and shTP53-EVs bound to polystyrene microspheres and stained for the indicated proteins or the correspondent isotype control (1 representative experiment of 3). (B) Alemtuzumab-mediated ADCP assay with normal hMB cells and J774A.1 macrophages in the presence of EVs derived from shCTRL and shTP53 hMB cells that were incubated (10 minutes) with anti–PD-L1 antibody (atezolizumab) prior to their addition to the coculture. The same concentration of anti–PD-L1 antibody diluted in PBS was used as control. Data shows 1 representative experiment of 3, with 5 replicates. (C) Immunoblotting detection of p53 and PD-L1 protein in shTP53/PD-L1–WT and -KO hMB cells. The corresponding total protein staining of the samples as a protein-loading control is shown. (D) Alemtuzumab-mediated ADCP assay with normal hMB cells with J774A.1 macrophages in the presence of shTP53/PD-L1–WT and -KO EVs; PBS was used as control. Data shows 1 representative experiment of 3, with 5 replicates. (E) Flow cell cytometer detection of PD-L1 in shCTRL and shTP53 hMB cells, treated with mafosfamide (CTX) or vehicle for 24 hours. Left panel shows half offset histogram representation of 1 representative experiment. Right panel displays PD-L1 protein expression percentage from 6 independent experiments. (F) Anti-CD20 (18B12)-mediated ADCP of shCTRL and shTP53 hMB cells pretreated or not with mafosfamide and cocultured with peritoneal macrophages exposed to anti-PD1 antibody (GS-696882) from 4 hours prior to the assay. Data shows 1 representative experiment of 3, with 5 replicates. (G) Survival curve of NSG mice IV injected with shTP53 hMB tumor cells treated IP with cyclophosphamide and alemtuzumab (CA) or CA and anti-PD1 antibody (GS-696882). PBS was used as control treatment (n = 12-18). (H) Alemtuzumab-mediated ADCP of different shTP53/PD-L1–WT and -KO clones coculture with J774A.1 macrophages (n = 3). (I) Number of mafosfamide (CTX) pretreated GFP+ hMB cells (shTP53/PD-L1–KO, shTP53/RAB27A-KO, and the corresponding shTP53 empty vector) remaining in the coculture with J774A.1 macrophages after ADCP assay performed in combination or not with alemtuzumab and normalized to the amount of hMB cells in a hMB single culture. Data shows 1 representative experiment, with 5 replicates. (J) Survival curve of NSG mice IV injected with shTP53/PD-L1-WT or shTP53/PD-L1-KO hMB tumor cells and IP treated after 10 days with cyclophosphamide and alemtuzumab (CA) or PBS as control treatment (n = 3-6). *P < .05, **P ≤ .01, ***P ≤ .001, ****P ≤ .0001. IP, intraperitoneal.
Likewise, shTP53-dependent PD-L1 cell surface expression was addressed in the context of alkylating treatment in vitro (Figure 5E). We observed significantly higher PD-L1 protein expression in shTP53 cells than in control cells, with PD-L1 upregulation even slightly increased after chemotherapy exposure (Figure 5E). However, CD47 and CD200 expression showed unaltered expression in all conditions (supplemental Figure 5A). We could corroborate that primary TP53-deficient malignant B-cells also expressed higher PD-L1 protein levels than the respective controls, shown by analyzing samples from Eµ-TCL1 mice and from CLL patients (supplemental Figure 5D-E).
Cotargeting the PD1/PD-L1 axis in preclinical B-cell lymphoma models
This observation prompted us to evaluate the relevance of the PD1/PD-L1 axis in the resistance to CIT. We performed an ADCP assay in the presence of anti-PD1 antibody in combination with chemotherapy (Figure 5F). The addition of anti-PD1 antibody to the coculture significantly improved the ADCP of both shCTRL and shTP53 hMB cells. This finding was confirmed by using the Myd88 p.L252P-driven DLBCL model, where PD1/PD-L1 inhibition restored the resistance to chemotherapy observed on the ADCP of TP53-deficient tumor cells (supplemental Figure 5C). In addition, the use of the triple therapy approach (anti-PD1 antibody, cyclophosphamide, and alemtuzumab) in the humanized mouse model of DHL with TP53-deficient lymphoma cells revealed a significantly increased overall survival of the mice compared with cyclophosphamide/alemtuzumab combination therapy (CA median equals 11 plus or minus 0.54 days vs CA plus anti-PD1 median equals 13 plus or minus 1.40 days; P = .023) (Figure 5G). To clarify the role of PD1/PD-L1 axis in the abrogation of macrophage antitumor functions on TP53-deficient lymphomas, we analyzed the ADCP of shTP53/PD-L1–KO hMB cells (Figure 5H-I). We demonstrated that PD-L1 deletion favored the ADCP of TP53-defcient cells in vitro (Figure 5H), showing a higher phagocytic response of macrophages upon CIT (Figure 5I). Finally, we evaluated the role of PD-L1 in the resistance to chemotherapy in vivo by injecting shTP53/PD-L1–KO hMB cells into NSG mice (Figure 5J). Our results showed a significantly prolonged survival of shTP53/PD-L1–KO vs shTP53/PD-L1–WT lymphoma-bearing mice (PBS+PD-L1–KO median equals 10 plus or minus 0.55 days vs PBS+PD-L1–WT median equals 8 plus or minus 0.61 days; P = .007). Importantly, CIT response was highly effective in mice injected with shTP53/PD-L1–KO tumor cells (Figure 5J) (PBS+PD-L1–WT median equals 10 plus or minus 0.58 vs CA+PD-L1–KO median equals 14 plus or minus 1.67 days; P = .005). Thus, CIT treatment in combination with PD1/PD-L1 inhibition improved TP53-induced therapy resistance. In conclusion, the upregulation of PD-L1 on TP53-deficient tumor cells appears to be a resistance mechanism amenable for therapeutic targeting in B-cell malignancies in addition to CIT.
Discussion
The concept of p53 as the central node in the DDR and “guardian of the genome” was recently expanded toward cell-nonautonomous effects by modulation of the TME by p53-deficient cancer cells.34,35 The functional impact of p53 on the TME has been shown in several tumor models, with the recovery of its function leading to the restoration of immune surveillance in liver cancer.36,37 Here, we show that in B-cell lymphoma cells, TP53 serves as a regulatory switch of the function of macrophages in the lymphoma TME.
In previous work, we could determine the essential role of cytokine release from lymphoma cells upon alkylating chemotherapy for sensitization of macrophage effector functions as the synergistic mechanism in CIT. 2,6 Now we show that an intact DDR is required to activate this mechanism of synergy and that p53 function serves as a central node of the DDR to control resistance against CIT. Corresponding to this observation, loss or mutation of TP53 is the most important resistance mechanism to CIT in CLL.12,38,39
The regulatory effect of TP53-deficient lymphoma cells on macrophage effector function was caused by a profoundly elevated secretion of EVs and an increased PD-L1 expression. Interestingly, we identified a central connection of a p53-dependent release of EVs and the expression of PD-L1 on cells and functional impact of PD-L1 carried by EVs
Multiple mechanisms of controlling PD-L1 expression have been described in malignant cells. Likewise, TP53-dependent control of PD-L1 expression has been described in small-cell lung cancer cells, involving miRNA-34a regulation of the PD-L1 3′UTR.40,41 Furthermore, TP53 status has been hypothesized to predict response to checkpoint inhibitor therapy in cancer.42,43 The functional relevance of immune oncology drugs targeting the PD-1/PD-L1 axis have so far mainly been attributed to T-cell activation and adaptive immunity.44,45 However, there is similar evidence that the PD1/PD-L1 checkpoint regulates macrophages in the TME.46 We have now identified PD-L1 to inhibit macrophages to effectively engulf lymphoma cells in the therapeutic setting, pointing to PD-L1 as a novel resistance mechanism for CIT regimens.
In this line, expression of PD-L1 was described as an integral component in a biomarker score indicating poor response to CIT of lymphoma.47 In DLBCL, patients with high expression of PD-1/PD-L1 on T cells and macrophages had significantly poorer survival after R-CHOP.48 In line with our data, it has been recently shown that loss of TP53 increases PD-L1 in a murine lymphoma model of DLBCL, inducing immune evasion, which could be overcome with PD1-blockade.49
Importantly, our data indicates that PD-L1 dependent immune suppression may occur independently of direct cell-cell interactions. We observe the increase of released EVs exposing PD-L1 to mediate the suppression of phagocytic function of effector macrophages. Notably, we see this novel phenomenon both in DLBCL and CLL murine models with genetic dysfunction of TP53, as well as in CLL patient samples. Using genetic targeting of PD-L1 on EVs, applying EV-deficient cells and immune checkpoint inhibitor blockade of EVs, we provide direct evidence of the decisive functional role of PD-L1+ EVs in regulating macrophage effector function and treatment response to CIT.
TP53 has been described to regulate EV secretion.50 More specifically, it has been proposed that mutant TP53 colon cancer cells reprogram macrophages toward tumor-supporting macrophages via miRNA (miR)-1246 delivered by EVs.22 Colorectal cancer−derived EVs were described to regulate polarization of tumor-associated macrophages by the miR-145, which is transcriptionally induced by p53.51,52 However, we do not see significant alterations in the miRNA profiles due to TP53 loss.
As for the context of chemotherapy and DNA damage, EV-based metabolic reprogramming by tissue-infiltrating macrophages underlines the role of EVs in cellular crosstalk.53 Also, an increase of EV-shedding was demonstrated to promote melanoma growth after chemotherapy.54 Melanoma-derived exosomes were identified to carry the immune checkpoint ligand PD-L1 and thereby contribute to immunosuppression.55 Recently, follicular lymphoma–derived EVs have been identified to promote polarization of the bone marrow stromal cell niche and modulate the TME toward tumor supportive function.56 So far, no specific therapeutic concept has been identified that counteracts EV-mediated effects. However, immune checkpoint inhibitors may offer an opportunity to target EV-mediated checkpoint control. In line with our hypothesis, we could effectively improve CIT by using an anti–PD-L1 antibody, atezolizumab, thereby significantly enhancing macrophage-mediated phagocytosis of lymphoma cells.
The conclusions from our findings are manifold. First, the results support the clinical evaluation of CIT with checkpoint inhibitors in order to overcome PD-L1–associated resistance. This strategy seems promising, as a combination of chemotherapy with immune checkpoint inhibitors was recently reported to be safe and to yield complete response rates of 77% in a phase 2 trial combining pembrolizumab with R-CHOP.16 Moreover, the loss of p53 function should call for innovative, intensified therapeutic strategies in lymphoma, including immunotherapies and immune checkpoint inhibitors. Particularly, combinations of targeted therapies such as immune modulators or BTK- or BCL2 inhibitors should be examined with regard to EV and PD-L1–mediated therapeutic responses.8,29,57 Finally, the release of immunosuppressive EVs can be recognized as a novel resistance mechanism in TP53-mutated lymphoma that may need to be monitored in future clinical studies.
Acknowledgments
The authors are indebted to their patients who contributed tissue and blood samples to this study. The authors are grateful for technical assistance from Yvonne Meyer and Sophie Neuber, support from Katrin Reiners, and the CECAD imaging and animal facilities.
This work was supported by the German research foundation (DFG) KFO 286 and CRC1530. C.P.P. was supported by the ‘Foerderprogramm Nachwuchsforschungsgruppen NRW 2015–2021, CAP Program of the Center for Molecular Medicine Cologne, and a research grant by Gilead Sciences. H.C.R. was supported by the German-Israeli Foundation for Research and Development (I-65-412.20-2016), the Deutsche Forschungsgemeinschaft (KFO-286-RP2), the Deutsche Jose Carreras Leukämie Stiftung (R12/08), the Else Kröner-Fresenius Stiftung (EKFS-2014-A06 to H.C.R., 2016_Kolleg.19), the Deutsche Krebshilfe (1117240 and 70113041), and the German Ministry of Education and Research (BMBF e:Med 01ZX1303A). H.B. was supported by Wilhelm-Sander Foundation and by the German Research Foundation (DFG BR 4775/2-1).
Authorship
Contribution: E.I. and C.P.P. contributed to study design; E.I., D.V., B.S., J.L.N., V.B., N.N., J.S., M.F., T.G., S.B., D.B., L.L., M.M., R.B., O.M., R.N., M.K., G.K., H.B., N.L., E.M., and N.N. contributed to data analysis and acquisition; B.C., J.N., S.B., M.N., M.P., R.R.-R., and M.K. contributed to bioinformatic analyses; H.C.R., E.M., L.F., and G.K. contributed analytical tools; C.P.P., B.C., N.N., H.B., and M.H. contributed clinical samples and annotation; C.P.P. contributed to study supervision and funding; and E.I., D.V., S.B., and C.P.P. contributed to manuscript preparation.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Christian P. Pallasch, Department I of Internal Medicine, Center for Integrated Oncology (CIO), University Hospital Cologne, Kerpener Str 62, 50937 Cologne, Germany; e-mail: christian.pallasch@uk-koeln.de.
For original data, please contact christian.pallasch@uk-koeln.de. scRNA-seq data may be found in a data supplement available with the online version of this article. Proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE [1] partner repository with the dataset identifier PXD019687.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.