

In this issue of Blood, 1 use detailed mutational analysis of aged mice and transplantation to evaluate the mouse as a model of clonal hematopoiesis.
Should you be fortunate enough to enjoy a long life, there is a chance you will develop clonal hematopoiesis (CH), even if you never know about it. Over the course of our lives, long-lived cells within self-renewing tissues such as hematopoietic stem cells (HSCs) can acquire and accumulate somatic mutations. If these mutations confer a fitness advantage, these mutated HSCs will have an increased output compared with nonmutant cells, leading to CH. With each decade of age older than 40, the prevalence of CH increases so that, by age 70, 10% to 20% of individuals will have detectable CH.2-4 A restricted set of genes has been reported to be mutated in CH,4,5 which is also mutated in myeloid malignancies, hinting that there is a continuum in transformation.
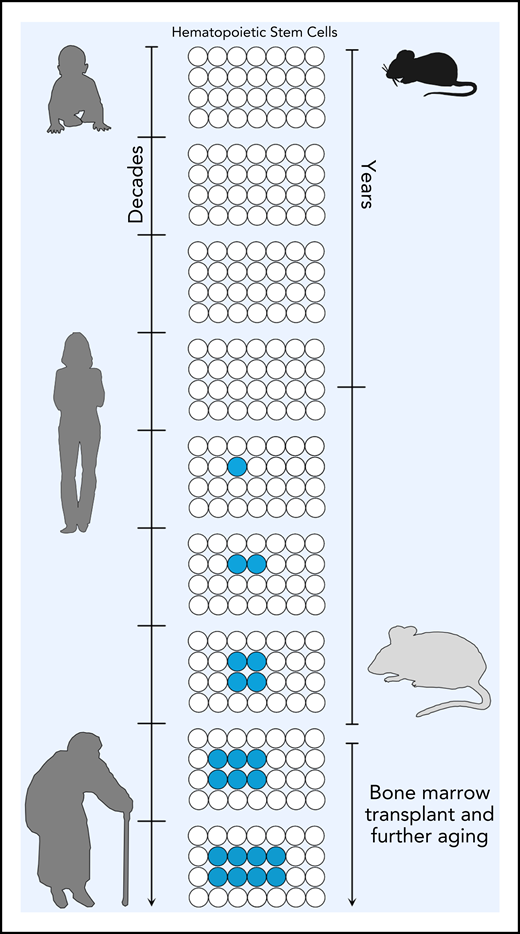
With age, hematopoietic stem cells can acquire somatic mutations (indicated in blue). If these mutations confer an advantage, these mutated HSCs can expand and give rise to clonal hematopoiesis. In humans, this has been documented from 40 years and older. As demonstrated by Chin et al, in mice, mutations in the same genes implicated in clonal hematopoiesis can be detected in 2-year-old mice (see figure). Upon secondary transplant, these preexisting HSCs can expand as seen in humans, demonstrating that mice can be used to model clonal hematopoiesis in vivo.
With age, hematopoietic stem cells can acquire somatic mutations (indicated in blue). If these mutations confer an advantage, these mutated HSCs can expand and give rise to clonal hematopoiesis. In humans, this has been documented from 40 years and older. As demonstrated by Chin et al, in mice, mutations in the same genes implicated in clonal hematopoiesis can be detected in 2-year-old mice (see figure). Upon secondary transplant, these preexisting HSCs can expand as seen in humans, demonstrating that mice can be used to model clonal hematopoiesis in vivo.
CH has been clearly demonstrated in humans, with analogous clonal hierarchies also detected in other aged tissues including the skin and esophagus.6 Our understanding of the evolution and natural history of CH has come from analysis of human samples, which have and will require decades to follow longitudinal cohorts for analysis. In the absence of time machines, analysis of human CH cannot be accelerated. This has raised interest in identifying animal models of CH. Mice have been the favored organism for the experimental manipulation and study of hematopoiesis for decades. Mice have a condensed lifespan relative to human (with a 2-year-old laboratory mouse approximating a 70-year-old human), have conserved genetic regulation of hematopoiesis compared with humans, and have an extensive history of genetic manipulation and longitudinal studies. Mice are not humans, however, and establishing the utility of mice to model CH is critical before a murine model can be used to explore and understand the clinical implications of CH.
Comparing 24-month-old C57BL/6 mice to 70-year-old humans, Chin et al demonstrate that, although mice have an 8.5-fold higher mutation rate per megabase of genomic DNA, human HSCs have an approximate 5-fold higher number of mutations than mouse. Despite these differences, both aged mouse and human HSCs show a conserved mutational signature dominated by the aging associated enrichment of C>T transitions in cytosine guanine dinucleotides. This demonstrates that core signatures of aging could be observed in aged murine HSCs. To directly address whether mutations arose in genes associated with CH in humans, the bone marrow of aged mice and from aged samples that had been transplanted into secondary recipients for a further 10 months was screened for a panel of genes known to account for most mutations in human CH (Dnmt3a, Tet2, Asxl1, Trp53, Sf3b1, Jak2, and Srsf2).2,4 From this analysis, using both digital droplet polymerase chain reaction and targeted exome sequencing, the authors report mutations in CH genes were present at a 2% frequency in aged mice. This increased to 18% in transplant recipients of the aged bone marrow, with evidence that preexisting mutant clones expanded from within the donor bone marrow after secondary transplant. The expansion of mutant clones after transplant has very recently been demonstrated in human HSCs, demonstrating that the aged murine model can recapitulate key features of human CH and HSC dynamics.7,8
The study by Chin et al establishes that murine HSCs acquire mutations in CH genes when aged and that these clones can expand further on transplantation. This demonstrates that murine HSCs are a tractable model to understand the evolution and dynamics of CH, offering a condensed lifespan and time frame to understand this process. The use of genetically modified mouse models with human-mimicking mutations in CH genes will be possible.9 This capability is directly relevant to understanding how these CH mutations impact normal hematopoiesis and provide a fertile environment for transformation. Additionally, the mouse will offer a platform to understand how extrinsic stressors, such as infection, cytokines, and cancer therapies, for example,10 contribute to the development of CH. The take-home message from this study is that patience is a virtue, at least when it comes to modeling CH.
Conflict-of-interest disclosure: The author declares no competing conflicts of interest.

