

In this issue of Blood, 1 identify the multifunctional protein pyruvate kinase muscle 2 (PKM2) in neutrophils as a key effector in stroke pathogenesis. The investigators show that induction of stroke results in nuclear translocation of PKM2 in neutrophils, driving a thrombo-inflammatory reaction through STAT3 signaling that exacerbates the severity of cerebral ischemia-reperfusion injuries.
Stroke is the fifth leading cause of death in the United States, with an estimated stroke-related death occurring every 4 minutes.2 The health care burden is substantial, with stroke accounting for an ∼$45 billion cost to the US health care system.2 The most significant problem for the clinical management of stroke is the limited number of treatment options. Recombinant-tissue plasminogen activator (r-tPA) is the only approved pharmacological therapy for stroke; however, its efficacy is limited by the narrow treatment window and a substantial risk for the development of a downstream intracranial hemorrhage.3 Mechanical thrombectomy is the only other treatment option for stroke but, similar to r-tPA treatment, roughly half of these patients suffer from suboptimal outcomes.4 A better understanding of mechanisms governing stroke pathogenesis, including the identification of key effector molecules, is a significant priority for improving its clinical management.
Pyruvate kinases are a family of proteins encoded by 2 genes that exist in 4 isoforms; they are expressed in almost all cell types.5 Dhanesha et al focused on PKM2 as a result of its high expression and upregulated activity in immune cells.6 PKM2 is unique among the family members in that it exists in 2 distinct forms in different intracellular compartments, including as a homotetramer in the cytoplasm and a homodimer in the nucleus. Intriguingly, the 2 forms seem to have distinct functions with the cytoplasmic tetramer acting in the final step of glycolysis to produce ATP and the nuclear dimer acting as a kinase that phosphorylates several targets to modulate the activity of transcription factors and cell cycle proteins.7 Interest in PKM2 has increased dramatically over the last several years. A need to better understand the complex cross talk between cell signaling and metabolic pathways has emerged as an important research focus across multiple fields, including cancer, inflammation, and immune regulation.
PKM2 has been implicated as an immune modulator capable of enhancing the activation status of multiple myeloid cell populations. Given the documented role of neutrophils and macrophages as key effector cells in stroke, this study centered on myeloid-derived PKM2 in pathogenic mechanisms in stroke and experimental cerebral ischemic injury. The investigators show that peripheral blood neutrophils (but not monocytes) isolated from stroke patients and mice challenged with cerebral ischemia exhibited increased nuclear PKM2 that was associated with elevated proinflammatory cytokine levels, an enhanced capacity to form neutrophil extracellular traps, and elevated expression of proinflammatory genes (eg, myeloperoxidase, elastase, HIF1α). A functional role for PKM2 was confirmed using a novel model to mediate selective deletion of PKM2 in myeloid-derived cells (ie, LysM-Cre/PKM2fl/fl mice), which ameliorated the peripheral neutrophil proinflammatory priming and hyperactivation status in mice challenged with cerebral ischemia-reperfusion.
The power and significance of the present study are derived from the depth of studies used in documenting a causative role for myeloid-derived PKM2 in the pathogenesis of experimental stroke. Indeed, the study by Dhanesha et al is an outstanding example of rigor and reproducibility in biomedical investigation. Conclusions are bolstered by multiple complementary analyses and strategies. For example, several clinically relevant experimental settings of stroke were analyzed, including mouse models using filament injury and clot injection/r-tPA models. Notably, validation analyses using multiple models is specifically recommended by the Stroke Therapy Academic Industry Roundtable guidelines for preclinical assessment of novel therapeutic targets.8,9 Studies are further strengthened by the consideration of comorbidities, such as hyperlipidemia (ie, using Apo−/− or Ldlr−/− mice), in the models, as well as sex as a biological variable by specifically analyzing male and female cohorts in separate experiments. Genetic deletion (ie, LysM-Cre/PKM2fl/fl mice) and pharmacological inhibition (ie, using the PKM2 nuclear translocation inhibitor ML265) intervention strategies were tested that collectively support the proposed mechanism and highlight potential translation. In total, the findings convincingly support the conclusion that PKM2 nuclear translocation in myeloid-derived cells (including neutrophils) exacerbates inflammation and tissue destruction following cerebral ischemia-reperfusion injury through a STAT3-dependent mechanism (see figure). Further, selective inhibition of PKM2 activity in myeloid cells has the potential to reduce brain infarct sizes, dampen cerebral inflammation, restore cerebral blood flow, and preserve neurocognitive function following stroke.
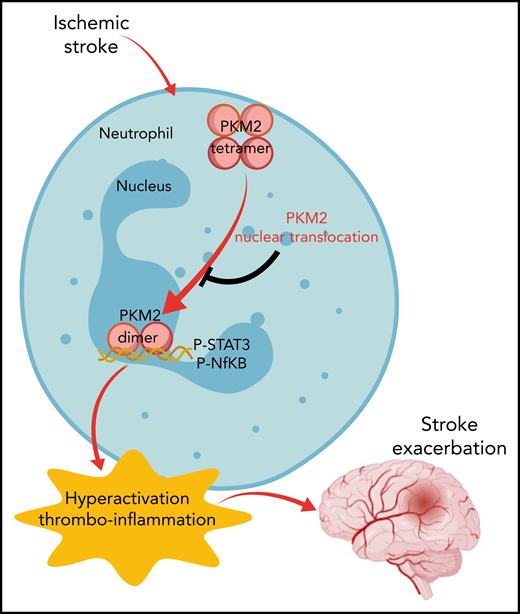
Model of neutrophil-derived PKM2 in the pathogenesis of cerebral ischemia-reperfusion injury. Upon neutrophil activation, PKM2 translocates to the nucleus as a homodimer to activate STAT3-dependent expression of proinflammatory genes. P-, phosphorylated.
Model of neutrophil-derived PKM2 in the pathogenesis of cerebral ischemia-reperfusion injury. Upon neutrophil activation, PKM2 translocates to the nucleus as a homodimer to activate STAT3-dependent expression of proinflammatory genes. P-, phosphorylated.
As is typically the case with studies highlighting novel mechanisms, new questions emerge. First, although the investigators provide compelling evidence that neutrophil-derived PKM2 is mediating the observed effects, it is possible that PKM2 expression in other cell types is contributing to stroke pathogenesis. In particular, additional studies should focus on other immune cell types, such as macrophages and T cells, which express PKM2 and have been linked to the pathogenesis of stroke and cerebral ischemia-reperfusion injury. Future studies should also clarify the precise PKM2 molecular mechanisms that exacerbate immune cell function in stroke. The investigators argue that nuclear PKM2 protein kinase activity is responsible for driving myeloid cell function in stroke. This conclusion is based on the efficacy of the inhibitor ML265 in limiting disease severity, which blocks nuclear translocation but not cytoplasmic glycolytic activity. However, considering that highly active inflammatory cells undergo a metabolic shift from oxidative phosphorylation to aerobic glycolysis, resembling the well described Warburg effect found in tumor cells, additional studies of PKM2 modulation of metabolic function is warranted. Indeed, accumulating studies support the concept that metabolic enzymes and their regulators, which are essential for the control of cellular metabolism, exert critical roles in regulating immune cell functions. Finally, if findings from the present studies are to be translated, it will be essential to determine the therapeutic window during which PKM2 inhibition is effective in reducing stroke pathologies and evaluate whether short- or long-term PKM2 inhibition is associated with deleterious effects within neuronal tissue or other organ systems.
Conflict-of-interest disclosure: The author declares no competing financial interests.

