Abstract
Disorders of coagulation, resulting in serious risks for bleeding, may be caused by autoantibody formation or by mutations in genes encoding coagulation factors. In the latter case, antidrug antibodies (ADAs) may form against the clotting factor protein drugs used in replacement therapy, as is well documented in the treatment of the X-linked disease hemophilia. Such neutralizing antibodies against factors VIII or IX substantially complicate treatment. Autoantibody formation against factor VIII leads to acquired hemophilia. Although rare, antibody formation may occur in the treatment of other clotting factor deficiencies (eg, against von Willebrand factor [VWF]). The main strategies that have emerged to address these immune responses include (1) clinical immune tolerance induction (ITI) protocols; (2) immune suppression therapies (ISTs); and (3) the development of drugs that can improve hemostasis while bypassing the antibodies against coagulation factors altogether (some of these nonfactor therapies/NFTs are antibody-based, but they are distinct from traditional immunotherapy as they do not target the immune system). Choice of immune or alternative therapy and criteria for selection of a specific regimen for inherited and autoimmune bleeding disorders are explained. ITI serves as an important proof of principle that antigen-specific immune tolerance can be achieved in humans through repeated antigen administration, even in the absence of immune suppression. Finally, novel immunotherapy approaches that are still in the preclinical phase, such as cellular (for instance, regulatory T cell [Treg]) immunotherapies, gene therapy, and oral antigen administration, are discussed.
Introduction
The formation of antidrug antibodies (ADAs) represents a major complication in the treatment of inherited coagulation diseases, while autoantibodies may also cause coagulation disorders. These humoral immune responses can result in a life-threatening risk of excessive bleeding, thus prompting advances in clinical immunotherapy for patients with bleeding disorders. The most extensive experience with immunotherapies for coagulation disorders is in hemophilia. Hemophilia A (HA) is an X-linked bleeding disease due to mutations in the F8 gene leading to factor VIII (FVIII) deficiency. Alloantibodies to the FVIII replacement therapy form in 30% of patients with severe disease (FVIII <1% of normal). These antibodies interfere with FVIII function and are referred to as inhibitors. In contrast, acquired hemophilia is caused by autoantibodies to FVIII that formed in a patient without hemophilia (Table 1). In replacement therapy for severe hemophilia B (HB), ∼3% of patients form inhibitors against factor IX (FIX, a serine protease whose enzymatic activity depends on its cofactor, FVIII).1 Three approaches to this problem have been developed (Figure 1): induction of antigen-specific immune tolerance (ITI), immune suppression therapy (IST), and restoring hemostasis through alternative molecules or pathways to circumvent the antibody blockade (such as bypass therapies, bispecific antibodies that mimic the function of FVIII or suppression of anticoagulant pathways). Antigen-specific ITI protocols were first used in the 1970s and showed that frequent IV delivery of FVIII can eliminate inhibitors.2 Furthermore, ITI protocols not only eradicate ADA but also promote lasting tolerance, serving as an important proof of principle that antigen-specific tolerance to the therapeutic drugs can be achieved in replacement therapy for genetic diseases. However, a preventative tolerance protocol is still lacking.
![Clinical immunotherapies and alternative approaches to eliminate or circumvent antibodies against coagulation factors formed in patients with bleeding disorders. These concepts are illustrated using inhibitor formation in the treatment of hemophilia as an example. Alternative strategies include [1] immune tolerance induction (ITI) by frequent IV administration of antigen; [2] transient immune suppression therapy (IST); and [3] restoration of hemostasis using nonfactor therapeutics, thus avoiding the use of the target antigen. Center: Inhibitory antibodies are produced by B cells upon their T helper cell-dependent activation. T follicular helper cells promote the organization of germinal centers and the production of antibodies. B cells may differentiate into memory B cells (BM) or plasma cells (PCs), which produce antibodies long-term. Ultimately, inhibitors prevent blood clot formation by reducing/eliminating the activation of factor X. This reaction is a critical component of the coagulation cascade and normally occurs through the cooperation of activated FVIII (FVIIIa) and factor IX (FIXa). Left: Although little is known about the mechanisms by which ITI reverses inhibitor production upon frequent IV factor administration, evidence supports that high antigen doses can directly inhibit BM. IST may use small molecule drugs such as cyclophosphamide, a DNA alkylating agent that eliminates proliferating cells such as activated B and T cells. Rituximab (anti-CD20 antibody) depletes mature B cells (but not BM or PCs). Regulatory T cells (Tregs) are able to suppress inhibitor formation. Therefore, experimental approaches use inhibition of the mTOR pathway with rapamycin/sirolimus (which aids in deletion of effector T cells, suppresses germinal center formation, and promotes Treg induction); oral antigen administration or hepatic gene transfer to induce Tregs, or Treg cell therapy, among other approaches. Right: Alternatively, nonfactor therapies bypass the effect of inhibitors by using a bispecific antibody that partially mimics the function of FVIII but is not recognized by FVIII inhibitors (a treatment that, however, only applies to HA) or promotes coagulation by elimination/neutralization of critical components of anticoagulant pathways (such as AT or TFPI) through small interfering RNA or monoclonal antibody therapy. AT, antithrombin III; TFPI, tissue factor pathway inhibitor.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/140/10/10.1182_blood.2022016530/3/m_bloodbld2022016530cf1.png?Expires=1769323501&Signature=rxX4mDQAbYQUvs5dHAXWY8IORRolQEbRcedQhZQ1IYkd58qlu-vgdw9qbUvDAcv6ljZuhhIbAmGzsTjj7geVHk2JqFzJuBJLoFknokcR5Z2VF4ntNDw5rSYTfwlcPJEeQT7RPxdwY-eg8JWFHY9DHwxxh~~8ukas1RKW0E6aLtIy4JpZmznG1kpYd4lfyZ0zQFuIlKBr5dmyCCh5FGQ-2EGPLnNZ1nBpekANxdeK43W1xU1Kqtbw834hn-6TdcpAfCF9Fh4CD~obsJSt8EuINVX37b-7LDgXtaOLPo-trLBdHUd2GT~THbZHxKl1K~AYtoRTNxYzlrrVyvXtdhXkMQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Clinical immunotherapies and alternative approaches to eliminate or circumvent antibodies against coagulation factors formed in patients with bleeding disorders. These concepts are illustrated using inhibitor formation in the treatment of hemophilia as an example. Alternative strategies include [1] immune tolerance induction (ITI) by frequent IV administration of antigen; [2] transient immune suppression therapy (IST); and [3] restoration of hemostasis using nonfactor therapeutics, thus avoiding the use of the target antigen. Center: Inhibitory antibodies are produced by B cells upon their T helper cell-dependent activation. T follicular helper cells promote the organization of germinal centers and the production of antibodies. B cells may differentiate into memory B cells (BM) or plasma cells (PCs), which produce antibodies long-term. Ultimately, inhibitors prevent blood clot formation by reducing/eliminating the activation of factor X. This reaction is a critical component of the coagulation cascade and normally occurs through the cooperation of activated FVIII (FVIIIa) and factor IX (FIXa). Left: Although little is known about the mechanisms by which ITI reverses inhibitor production upon frequent IV factor administration, evidence supports that high antigen doses can directly inhibit BM. IST may use small molecule drugs such as cyclophosphamide, a DNA alkylating agent that eliminates proliferating cells such as activated B and T cells. Rituximab (anti-CD20 antibody) depletes mature B cells (but not BM or PCs). Regulatory T cells (Tregs) are able to suppress inhibitor formation. Therefore, experimental approaches use inhibition of the mTOR pathway with rapamycin/sirolimus (which aids in deletion of effector T cells, suppresses germinal center formation, and promotes Treg induction); oral antigen administration or hepatic gene transfer to induce Tregs, or Treg cell therapy, among other approaches. Right: Alternatively, nonfactor therapies bypass the effect of inhibitors by using a bispecific antibody that partially mimics the function of FVIII but is not recognized by FVIII inhibitors (a treatment that, however, only applies to HA) or promotes coagulation by elimination/neutralization of critical components of anticoagulant pathways (such as AT or TFPI) through small interfering RNA or monoclonal antibody therapy. AT, antithrombin III; TFPI, tissue factor pathway inhibitor.
Clinical immunotherapies and alternative approaches to eliminate or circumvent antibodies against coagulation factors formed in patients with bleeding disorders. These concepts are illustrated using inhibitor formation in the treatment of hemophilia as an example. Alternative strategies include [1] immune tolerance induction (ITI) by frequent IV administration of antigen; [2] transient immune suppression therapy (IST); and [3] restoration of hemostasis using nonfactor therapeutics, thus avoiding the use of the target antigen. Center: Inhibitory antibodies are produced by B cells upon their T helper cell-dependent activation. T follicular helper cells promote the organization of germinal centers and the production of antibodies. B cells may differentiate into memory B cells (BM) or plasma cells (PCs), which produce antibodies long-term. Ultimately, inhibitors prevent blood clot formation by reducing/eliminating the activation of factor X. This reaction is a critical component of the coagulation cascade and normally occurs through the cooperation of activated FVIII (FVIIIa) and factor IX (FIXa). Left: Although little is known about the mechanisms by which ITI reverses inhibitor production upon frequent IV factor administration, evidence supports that high antigen doses can directly inhibit BM. IST may use small molecule drugs such as cyclophosphamide, a DNA alkylating agent that eliminates proliferating cells such as activated B and T cells. Rituximab (anti-CD20 antibody) depletes mature B cells (but not BM or PCs). Regulatory T cells (Tregs) are able to suppress inhibitor formation. Therefore, experimental approaches use inhibition of the mTOR pathway with rapamycin/sirolimus (which aids in deletion of effector T cells, suppresses germinal center formation, and promotes Treg induction); oral antigen administration or hepatic gene transfer to induce Tregs, or Treg cell therapy, among other approaches. Right: Alternatively, nonfactor therapies bypass the effect of inhibitors by using a bispecific antibody that partially mimics the function of FVIII but is not recognized by FVIII inhibitors (a treatment that, however, only applies to HA) or promotes coagulation by elimination/neutralization of critical components of anticoagulant pathways (such as AT or TFPI) through small interfering RNA or monoclonal antibody therapy. AT, antithrombin III; TFPI, tissue factor pathway inhibitor.
Here, we review clinical immunotherapies for inherited and acquired hemophilia, von Willebrand disease (VWD), and other rarer bleeding disorders, using alternative strategies to treat the disease that avoid the use of the target antigen or immune suppression, as well as innovative new approaches in early clinical and preclinical stages. The availability of animal models that recapitulate antibody formation against coagulation factors enables the current development of next-generation immunotherapies. These approaches also take advantage of recent advances in immunotherapies for cancer and inflammatory autoimmune disease that have seen a revolution through engineered T cell and monoclonal antibody therapies, among other technologies.3
Immune modulatory strategies for alloantibodies to FVIII in HA
FVIII protein replacement is effective in preventing and controlling bleeding, and a range of plasma-derived and recombinant FVIII molecules are available in the clinic. However, inhibitor formation substantially raises the risk for morbidity and mortality in patients.4 High-titer inhibitors (>5 Bethesda Units, BU/mL; 1 BU is defined as the reciprocal of the dilution of test plasma in which 50% of FVIII activity is inhibited) present the most challenging clinical scenario since FVIII therapy is no longer effective. Even in the presence of low-titer inhibitors, FVIII replacement at much higher doses (three to fivefold) than standard therapy is needed to provide effective hemostasis.
To date, the only widely used efficacious strategy for inhibitor eradication and restoring hemostatic response to FVIII is ITI.2,5,6 Over 40 years, the outcomes of ITI were based on noncontrolled, relatively smallscale studies with variable follow-up. Then in 2012, a multicentric, randomized, prospective controlled study compared high-dose (HD) vs low-dose (LD) FVIII in an international ITI study based solely on the evaluation of “good prognosis patients” (Tables 1 and 2)7 improved our understanding of ITI efficacy. An approximately 70% success rate was found in both HD and LD cohorts during a 3-year follow-up. The time for inhibitor eradication and normal FVIII pharmacokinetics was faster in the HD cohort, while in the LD cohort bleeding episodes were approximately threefold higher. Thus, evidence-based ITI data favored HD FVIII.
The role of IST coupled with ITI
IST is not part of the standard ITI studies, which are often restricted to “good prognosis patients.” Therefore, most studies of IST are on patients with poor prognosis (Table 2), and there are no specific guidelines for the management of this challenging group of patients. These studies are complicated by multiple factors such as age, duration of previous inhibitor titers, prior attempts of ITI, bleeding phenotype, and comorbidities. It has not been possible to define an optimal IST regimen as an adjunct therapy for ITI. Not unexpectedly, there are no randomized studies on the use of IST in inhibitor patients undergoing ITI. Notably, IST without ITI is often ineffective for sustained inhibitor eradication. The recent approval of emicizumab for prophylaxis in HA with inhibitors that failed ITI or are of poor prognosis opens a new area of investigation that is not without limitations and safety considerations (see below). Future studies will be needed to address some of these questions.
Data on the most common immunosuppressive drugs (the DNA alkylating agent cyclophosphamide and the B-cell–depleting monoclonal antibody rituximab) used in distinct ITI strategies were recently reviewed.8-10 Some other IST drugs have also been tested but showed inferior efficacy when used alone (ie, not combined with ITI; albeit combinations such as rituximab and the mTOR inhibitor sirolimus may be effective).11 However, there is no consensus about the reporting of outcomes or follow-up in these studies. Outcomes of ITI can be defined as (1) complete response (CR) (negative inhibitor titer and normalization of FVIII recovery and half-life with normalized pharmacokinetics [PK]); (2) partial response (PR) (initial inhibitor titer negative but with abnormal PK); (3) relapse (inhibitor recurrence and abnormal PK); and (4) failed response (FR) (inhibitor failed to decline by >20% throughout the regimen and no partial response observed).
In adult patients with HA, cyclophosphamide was effective alone or in combination with other drugs in ∼44% of cases when coupled with ITI.9,10 In addition, IST drug combinations were associated with reduced numbers of FR. In children, ITI coupled with cyclophosphamide resulted in CR of 41%; there was also an increase in the CR when other immunosuppressants were added. However, cyclophosphamide is no longer used in pediatric patients. Finally, IV immune globulin (IVIG) combined with cyclophosphamide seems to provide a modest further improvement in both pediatric and adult inhibitor patients.9
The advent of rituximab (a chimeric murine/human antibody against CD20) generated great interest for use in ITI. Rituximab depletes peripheral B cells (>90%) but has no effect on memory B, plasma, and T cells.12,13 The rates of CR in small series of adult and pediatric populations were 61% and 40%, respectively. Not surprisingly, the efficacy of rituximab without ITI is modest or even poor. Moreover, in a phase 2 prospective study, using rituximab alone to prevent an anamnestic response to FVIII was effective in only 19% of cases.14
IST in nonsevere HA (NSHA) patients
The development of inhibitors in NSHA increases morbidity by exacerbating the bleeding episodes when FVIII levels are decreased to <1%. Retrospective analyses show that clinically relevant inhibitors affect 3% to 13% of NSHA.15,16 The management of these patients is challenging because of the often older age at the time of inhibitor onset. However, >70% of inhibitors resolve spontaneously, although anamnestic responses remain a problem. Various combinations of ITI and IST have been effective in >70% of cases.16 IST alone is sometimes preferable in older patients. For example, rituximab was found to be effective in some patients, albeit experience is limited.17-19
IST in treatment of acquired HA
Acquired HA (AHA) results from the development of inhibitory autoantibodies that neutralize FVIII but show major differences to FVIII alloantibodies in congenital HA (Table 1). AHA has an incidence of 1 to 1.5 cases per million per year, and thus, diagnostic awareness and initial treatment are sometimes delayed.20,21 In 50% of cases, the disease is idiopathic (Table 1). Clinically, AHA is characterized by new onset bleeding most often into soft tissues, including muscle, skin, and gastrointestinal and genitourinary tracts in >85% of patients. In contrast to X-linked HA, the presence of hemarthrosis is uncommon. Although spontaneous remission of the disease occurs, prolonged duration is associated with high morbidity and mortality rates.22-24
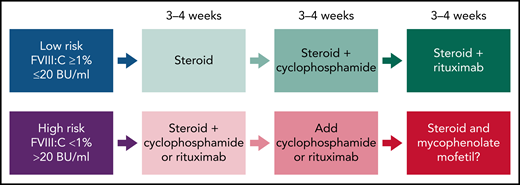
Recent guidelines in AHA (Figure 2) suggest that for the low-risk group (FVIII >1% and <20 BU/mL), treatment could start with steroids alone, and if there is no inhibitor resolution after 3 to 4 weeks, cyclophosphamide is added.22-24 In the absence of response, more recently, rituximab has replaced cyclophosphamide. In the high-risk group (FVIII <1% and >20 BU/mL), combination therapy (steroids coupled with cyclophosphamide or rituximab) is used initially for 3 to 4 weeks to achieve complete remission and to accelerate and sustain inhibitor eradication.22-24 In the case of failure, IST regimens containing mycophenolate mofetil (MMF) could be effective. However, the evidence is limited, and the duration of MMF is variable.25 Alternatively, in the case of contraindication to steroids, rituximab could be the first choice. It is important to consider the time of IST required for inhibitor eradication. In the high-risk group, early initiation of more intensive IST for a shorter period of time may be more attractive than delayed initiation/duration of IST because of the increased risk of infection.26
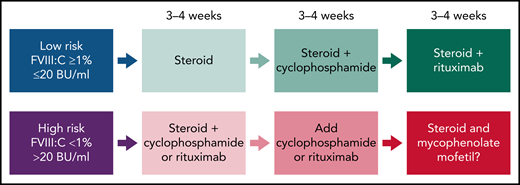
Overall strategy for IST for acquired HA. Proposed therapies are based on prognostic risk factors (FVIII clotting activity and inhibitor titers in Bethesda Units, BU). The response could be assessed as a complete response or significant and sustained improvement on the FVIII:C and/or reduction of inhibitor titers at the end of 3 to 4 weeks. In the case of failure in achieving these outcomes, the therapeutic intervention should proceed with the subsequent regimen.
Overall strategy for IST for acquired HA. Proposed therapies are based on prognostic risk factors (FVIII clotting activity and inhibitor titers in Bethesda Units, BU). The response could be assessed as a complete response or significant and sustained improvement on the FVIII:C and/or reduction of inhibitor titers at the end of 3 to 4 weeks. In the case of failure in achieving these outcomes, the therapeutic intervention should proceed with the subsequent regimen.
Mortality in AHA is estimated to be ∼20% in patients >65 years and is often related to the underlying cause of the disease.26-29 However, the risk of infection because of the duration of IST and the advanced age of patients is of major concern. Infection (∼15%) and potential cardiovascular prothrombotic morbidity are the leading causes of mortality in AHA rather than the bleeding itself, which is now likely responsible for <5% of deaths.
Approximately 7% to 21% of AHA cases are because of postpartum inhibitor formation, occurring with an incidence of ∼1 in 350 000 births. Bleeding episodes are noted in the first 3 months but may occur up to 1 year after delivery.30-32 The bleeding phenotype is comparable to AHA of other causes, in addition to vaginal/uterine hemorrhage. There is also a potential for transplacental transfer of the FVIII antibodies, resulting in a risk for neonatal bleeding.33 Spontaneous remission has been observed but can take several months.31,34 Thus, IST is indicated to prevent bleeding episodes, and 70% of women are treated with steroids alone (cytotoxic-based therapy is mostly avoided because of breastfeeding and fertility concerns). Rituximab and IVIG have been used for those cases that failed on steroids.23,31,35
Treatment of inhibitors in HB
The formation of inhibitors to FIX in HB is a rare complication affecting 1% to 3% of all patients and ∼5% of those with severe disease.36,37 In addition, systemic complications such as allergic and anaphylactic reactions to FIX protein can accompany or follow the development of inhibitors to FIX. In several studies with limited numbers of patients, the success rates of ITI are low (∼20%).36,38 The development of nephrotic syndrome is likely to adversely impact ITI. Astermark and colleagues reported recently that multiple attempts of ITI are feasible and efficacious.38 IST is often coupled with ITI, although the choice of drugs varies widely.
Antibody- and small interfering RNA (siRNA)-based treatments as alternatives to traditional immunotherapy
Alternatives to immunotherapy are to either promote coagulation through pathways that do not require FVIII or FIX or to use alternative molecules, thereby avoiding the antigen that is targeted by the inhibitors. The former involves siRNA and monoclonal antibody therapies currently in clinical development to suppress anticoagulant molecules such as antithrombin and tissue factor pathway inhibitor, respectively. This strategy can restore hemostasis in patients with inhibitors against FVIII or FIX.39 Treatment of HA has been dramatically changed by the development of the bispecific humanized immunoglobulin G4 (IgG4) monoclonal antibody emicizumab that is given subcutaneously and partially mimics the function of activated FVIII by binding to FIX and factor X. Importantly, emicizumab is not recognized by FVIII inhibitors and is now licensed for prophylactic use to prevent bleeds in HA patients with and without inhibitors.39,40 Overall, breakthrough bleeds still occur with these therapies, and FVIII infusions will still be required, at least transiently, thus posing a risk for anamnestic responses to FVIII. The use of concomitant replacement therapy or bypass products has imposed safety limitations such as the development of thrombotic complications that cannot be predicted with certainty.
Immunotherapies in the management of VWD and immune responses to other coagulation factors
Immune reactions against von Willebrand factor (VWF) occur significantly less frequently than against FVIII. The first report of a VWF alloantibody was >40 years ago, and since then, many single case reports and a few multicenter surveys have been published.41,42 Most studies indicate that the prevalence of anti-VWF alloantibodies in patients with severe VWD is between 5% and 10% (Table 3).43 Alloantibody development has been documented almost exclusively in patients with type 3 VWD, although rare case reports of alloantibodies in other VWD subtypes have been reported.44 Thus, this immunologic complication is a rare finding in a rare disease, a phenomenon that has significantly limited the acquisition of knowledge concerning pathobiology and clinical management.
The pathogenetic mechanisms of anti-VWF alloantibody development are not well defined. There is an apparent genetic risk component, with the earliest cases of alloantibody generation reported in patients with null mutations, either deletions or stop codons.45,46 However, no systematic genotype/phenotype analysis has been conducted, and no other nongenetic risk factors have been identified.
In the limited characterization of VWF alloantibodies, they have been shown to be polyclonal and of various IgG subclasses but with a predominance of IgG4, a feature also seen with anti-FVIII inhibitors.47 While IgG4 antibodies do not activate complement, anti-VWF alloantibodies have been shown to possess precipitating properties in some instances, and at high titers, they can also result in anaphylactic/anaphylactoid reactions upon infusion of VWF concentrates. The basis of these reactions is poorly understood. The literature contains 1 case where there was evidence of complement activation and another in which there was indirect evidence of an IgE-mediated acute hypersensitivity reaction.48,49
The epitope specificity of the alloantibody response has not been sufficiently examined to define immunodominant regions of VWF. Nevertheless, a recent phage display-based examination confirmed the polyclonal nature of the antibody response, hinted at the likelihood of conformational epitopes, and demonstrated very similar patterns of epitope specificity in 2 brothers possessing the same large VWF deletion.50
The presence of VWF alloantibodies is usually detected through a lack of clinical response to therapy. Unlike in hemophilia, where screening for inhibitory antibodies is often performed in patients receiving frequent treatment, laboratory testing for VWF alloantibodies is rarely conducted. Patients showing a poor clinical response to VWF replacement therapy should undergo postinfusion recovery and half-life analysis, and in the laboratory, mixing studies with normal plasma with subsequent quantification of platelet, collagen, and FVIII binding function of VWF assessed.51 There is no evidence that VWF alloantibodies are time-dependent in their inhibitory activity. An absence of functional inhibition does not exclude the presence of nonneutralizing antibodies that bind VWF and accelerate its clearance. Detection of these antibodies can be achieved with a solid phase binding assay with the caveat that false positive results can be obtained if appropriate control samples are not included.
An important consideration in the identification and characterization of anti-VWF alloantibodies is that routine testing for this complication is uncommon. Thus, any data available for this treatment complication will not involve a formally standardized testing strategy and suggests that testing should preferably be conducted in one of the few reference laboratories accustomed to performing these analyses.
There is no clearly effective strategy for the management of anti-VWF alloantibodies. The literature contains infrequent case reports using recombinant FVIII, rFVIIa, and platelets to treat bleeding, and recently the FVIII mimetic emicizumab has been used in this context.52 Remarkably, almost nothing is known about the potential of immunotherapies for this condition. There is a single case report of successful immune tolerance in a child with anti-VWF alloantibodies but no other objective evidence of success with immune modulatory therapies.53
While the incidence of alloimmunization to VWF in severe VWD is rare, there is a sense that the incidence of acquired von Willebrand syndrome (aVWS) is increasing, in part because of enhanced recognition but also because of a growing elderly population.54 The pathobiology of aVWS involves a range of mechanisms, a minority of which relate to the generation of anti-VWD antibodies. These antibodies can accompany lymphoproliferative disorders, including monoclonal gammopathies of uncertain significance and autoimmune diseases. When they are present, they will usually result in low plasma concentrations of VWF, presumably because of an accelerated clearance mechanism.55
Other aVWS-associated pathogenetic mechanisms also involve more rapid clearance of VWF and include the adhesion of VWF to tumor cell surfaces and VWF proteolysis in a variety of high-shear cardiovascular disorders (eg, aortic stenosis and treatment of ventricular assist devices).56,57 In a registry of 186 aVWS cases, 48% were associated with lymphoproliferative disorders, 21% with cardiovascular disease, 15% with myeloproliferative conditions, 5% with other cancers, and 2% with miscellaneous immunologic conditions.55
As all these immune and nonimmune pathophysiologies are rare, controlled data on treatment outcomes are lacking. In some instances, such as the cases of aVWS associated with high-shear cardiovascular disorders, correction of the primary condition will usually correct the VWF pathology. However, in the other pathophysiologic states, an assortment of case reports and small case series suggest that desmopressin (DDAVP), FVIII/VWF concentrates, IVIG, and immunosuppression have all been shown to produce some benefit in ∼30% of cases.58
In addition to the characterization of the immune responses to FVIII, FIX, and VWF, there is also a recognition that rare allo- and autoimmune responses can occur against most of the procoagulant proteins.59 In a study of 118 FXI-deficient patients, inhibitory antibodies were documented in 7 cases, all homozygous for the same null variant (Glu117Stop) and all of whom were treated with plasma infusions.60 Acquired anti-FV antibodies have been reported in the past, after exposure to FV-containing topical bovine thrombin preparations and in association with other comorbidities in elderly patients.61,62 Acquired FX deficiency can develop either through adsorption of the protein on amyloid aggregates or in association with anti-FX autoantibodies.63 Lastly, acquired FXIII autoantibodies have recently been identified with increasing frequency.64 These anti-XIII autoantibodies are found most frequently in elderly patients and are often associated with severe bleeding symptoms, including intracranial bleeds. As with inherited FXIII deficiency, routine hemostasis tests are normal in these cases, and confirmation of an anti-FXIII antibody requires specific assays to evaluate FXIII functional neutralization and/or binding to FXIII. All these conditions, and other rare bleeding disorder complications, are exceedingly infrequent and thus very poorly characterized in terms of both pathophysiology and clinical management.
Preclinical development of next-generation immunotherapies for bleeding disorders
Despite its success, traditional ITI to eradicate antibodies against coagulation factors in the treatment of hemophilia has multiple limitations, which include the need for frequent IV infusions over months, high costs, ineffectiveness in some patients, serious complications with FIX inhibitors, and lack of a prophylactic/preventative protocol. IST bears risks of infection, and nonfactor therapies may not be universally applicable and may bear thrombotic risk. These concerns sparked the development of a plethora of preclinical studies to explore alternative approaches, taking advantage of advances in immunotherapy, improved understanding of immune regulation, and novel technologies for effective drug and antigen delivery.65,66 While a comprehensive review is beyond the scope of this article, we will highlight several recent innovative strategies.
Once it was recognized that CD4+CD25+FoxP3+ regulatory T cells (Tregs) are able to control inhibitor formation against coagulation factors, diverse strategies have been employed to generate Treg-dominated responses in vivo or to develop cellular immunotherapies using gene-modified T cells, which has revolutionized the treatment of blood cancers.67 It is, therefore, logical that analogous tolerogenic lymphocyte therapies could be developed (Figure 3). Notably, ex vivo expanded autologous polyclonal Tregs suppressed FVIII inhibitor formation in mice.68 Unresponsiveness extended beyond the persistence of transplanted Tregs, which aid in the induction of endogenous antigen-specific Tregs through conditioning of dendritic cells. However, high cell doses are required. A potentially superior approach is to use gene transfer to redirect antigen specificity.
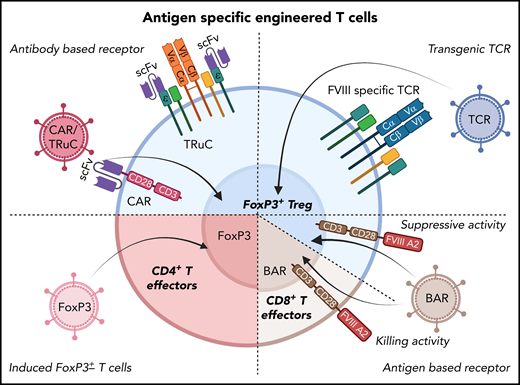
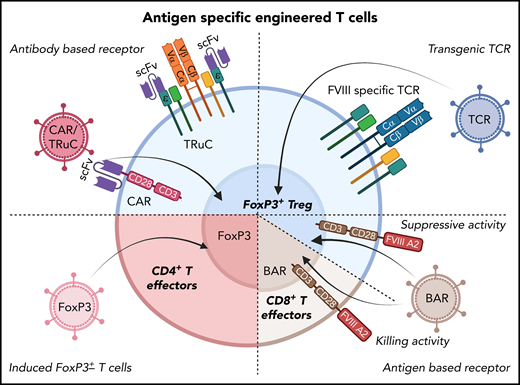
Potential cellular immunotherapies to suppress inhibitor formation in factor replacement therapy for hemophilia based on the engineering of T cells that have emerged from preclinical studies. In preclinical studies, T cells were gene-modified ex vivo by viral gene transfer, expanded, and subsequently transplanted to suppress inhibitor formation. These approaches seek to generate antigen-specific Tregs, as shown in several proof-of-principle studies on FVIII. Examples include redirection of antigen-specificity by transduction of CD4+FoxP3+ Tregs with FVIII-specific chimeric antigen receptor or T-cell receptor fusion construct (TRuC), which avoids major histocompatibility complex restrictions (top left); or with FVIII-specific TCR (top right). B-cell antigen receptors (BARs) use a portion of FVIII so that gene-modified CD4+FoxP3+ Tregs suppress FVIII-specific B cells (which express B-cell receptors for FVIII; bottom right). Alternatively, the introduction of a BAR to CD8+ T cells enables these cytolytic cells to eliminate FVIII-specific B cells (bottom right). Finally, expanded FVIII-specific effector CD4+ T cells can be reprogrammed to become Tregs by FoxP3 gene transfer (bottom left).
Potential cellular immunotherapies to suppress inhibitor formation in factor replacement therapy for hemophilia based on the engineering of T cells that have emerged from preclinical studies. In preclinical studies, T cells were gene-modified ex vivo by viral gene transfer, expanded, and subsequently transplanted to suppress inhibitor formation. These approaches seek to generate antigen-specific Tregs, as shown in several proof-of-principle studies on FVIII. Examples include redirection of antigen-specificity by transduction of CD4+FoxP3+ Tregs with FVIII-specific chimeric antigen receptor or T-cell receptor fusion construct (TRuC), which avoids major histocompatibility complex restrictions (top left); or with FVIII-specific TCR (top right). B-cell antigen receptors (BARs) use a portion of FVIII so that gene-modified CD4+FoxP3+ Tregs suppress FVIII-specific B cells (which express B-cell receptors for FVIII; bottom right). Alternatively, the introduction of a BAR to CD8+ T cells enables these cytolytic cells to eliminate FVIII-specific B cells (bottom right). Finally, expanded FVIII-specific effector CD4+ T cells can be reprogrammed to become Tregs by FoxP3 gene transfer (bottom left).
Initial success with this approach in preclinical models was achieved by the transfer of a T-cell receptor (TCR) gene to human Tregs.69 This TCR with specificity to an epitope of the C2 domain of human FVIII had previously been cloned from a HA patient with an inhibitor response. However, a variety of TCRs would have to be used because of differences in HLA. Thus, to eliminate HLA restrictions, Tregs were engineered to express chimeric antigen receptors (CARs). These are comprised of an antibody-derived single chain variable fragment (ScFv), displayed on the cell surface to bind to the target antigen, a transmembrane region, intracellular signaling sequences to provide costimulation, and the CD3ζ chain of the TCR. Gene-modified human Tregs expressing a CAR for the A2 domain of human FVIII (on the basis of an ScFv isolated from a phage display library) potently suppressed inhibitor formation.70 Similarly, murine Tregs modified to express FVIII-specific CARs and FoxP3 transgenes were suppressive in HA mice.71 However, when a high-affinity CAR construct was used that included an ScFv from a patient-derived FVIII inhibitor, Treg engagement surprisingly induced a robust effector phenotype that was distinct from the activation signature of natural Tregs.72 In contrast, complexing TCR-based signaling with the identical ScFv recognition in the form of a TCR fusion construct (TRuC) Treg resulted in controlled antigen-specific signaling via engagement of the entire TCR complex and suppression of inhibitor formation.72 Alternatively, one can generate chimeric receptors that display a portion of the FVIII antigen rather than an antibody binding site on the cell surface. Such a “B-cell antigen receptor” (or BAR) can then be introduced to endogenous Tregs or CD8+ T cells to target B cells with a B cell receptor to FVIII to suppress or kill these B cells, respectively.73-75
Another way to induce immune tolerance is to take advantage of natural tolerogenic microenvironments such as the liver and small intestine. For instance, adeno-associated viral (AAV) and lentiviral gene transfer to hepatocytes or liver sinusoidal epithelial cells have resulted in induction of immune tolerance to FVIII or FIX.76-79 Hepatic gene transfer in hemophilic mice and dogs was effective not only in prevention but also in the reversal of inhibitor formation and, encouragingly, the IgE-mediated anaphylactic phenotype in response to FIX.80 Robust antigen expression tilted the response to Treg induction and directly inhibited memory B cells.81 In an attempt to adapt oral immunotherapy (OIT) for hemophilia, chloroplast transgenic crop plants were produced for expression of FVIII or FIX antigens fused to a transmucosal carrier protein.82 Oral delivery of a freeze-dried powder of leaf cells allows for the cost-effective production of bioencapsulated antigen.82,83 Repeat delivery suppressed inhibitor formation against IV human FVIII and FIX in hemophilic animals,84-87 and also prevented anaphylaxis against FIX. The complex interleukin 10 (IL-10)-dependent tolerance mechanism includes, among other features, induction of FoxP3+ and LAP+ CD4+ Tregs.88-90
Advantages and limitations of new immunotherapy approaches
Treg engineering is currently still a specialized and expensive approach that may, however, become realistic in the future as experience grows and the technology is further advanced. The safety of hepatic gene transfer for hemophilia awaits further evaluation in humans (eg, in multiple ongoing phase 3 clinical trials using AAV vectors).91,92 It remains to be seen how effective any antigen-specific approaches are for inhibitor reversal in humans if immune-suppressive drugs are not included or whether at least transient IST is needed.
Compared with other approaches, plant-based oral tolerance may have fewer safety concerns for prophylactic tolerance applications in pediatric patients. Experience with OIT for food allergies shows that durable tolerance can be achieved if therapy is initiated early in life.82 Tolerance induction may even be possible in utero through transplacental transport of Fc-fusion antigens.93 Alternatively, FoxP3+ Tregs may be induced and empowered in vivo to induce tolerance using IL-2 muteins or IL-2/anti–IL-2 complexes.94,95
Summary and future outlook
The development of inhibitory antibodies to coagulation proteins has been recognized for several decades, but despite intensive investigation over an extended period of time, our understanding of the pathogenic mechanisms responsible for these disorders is still very poor. This challenge is exacerbated by the fact that these complications occur infrequently in rare (sometimes very rare) diseases. Nevertheless, successful immunotherapies based on ITI and/or IST have been developed in HA and acquired hemophilia. Moreover, the problem of treating inhibitor patients has now been elegantly addressed in HA with the bispecific antibody emicizumab, which has demonstrated excellent hemostatic activity in patients with alloantibodies against FVIII (but no clear beneficial effect on inhibitor titers).96,97 Other nonfactor therapies are in development to circumvent inhibitors against both FVIII and FIX in HA and B.
In our opinion, the induction of immune tolerance to the target coagulation protein is desirable, wherever possible, to enable unopposed substitution therapy for the prevention and/or treatment of bleeding. Presently, such an ITI approach is mostly restricted to HA. While no preventive tolerance protocols are available in the clinic, innovative and mechanistically better-defined tolerance approaches are in preclinical development in hemophilic animals. Ironically, the development of preventive immune tolerance to FVIII is complicated by the increased use of emicizumab, so patients are exposed to FVIII antigen only later in life and far less frequently. It remains to be seen if this reduces the overall incidence of inhibitor formation compared with FVIII replacement in previously untreated patients or perhaps increases the risk when FVIII is eventually given to restore hemostasis following trauma or during surgery.
In the case of autoantibodies against coagulation factors, IST is typically employed. However, IST is also an option in the X-linked hemophilias when ITI failed or had to be discontinued (as is often the case for FIX inhibitors) and for the rarer disorders when factor products or experience with immunotherapy are limited or lacking. Growing experience in hemophilia and other bleeding disorders with immune and alternative therapies could be instrumental in the development of analogous strategies for protein or gene-based treatments of many other hematological and nonhematological disorders that are complicated by adaptive immune responses.
Acknowledgments
The authors thank Jyoti Rana for help with Figure 3.
This work was supported by NIH grants U54 HL142012 to V.R.A., D.L., and R.W.H.; R01 HL131093 and P01HL160472 to R.W.H.; and R01 HL158781 to V.R.A.; by Indiana Collaborative Initiative for Talent Enrichment (INCITE) funds (provided by Lilly Endowment) and by the Riley Children’s Foundation to R.W.H.; a CIHR Foundation Grant FDN-154285 to D.L. D.L. is the recipient of a Canada Research Chair in Molecular Hemostasis.
We dedicate this article to Valder R. Arruda, who died during the writing of the manuscript.
Authorship
Contribution: V.R.A., D.L., and R.W.H. wrote the manuscript.
Conflict-of-interest disclosure: R.W.H. holds patents on oral tolerance for hemophilia, received grant support from Spark Therapeutics for gene therapy studies, and serves as a consultant for Intellia Therapeutics and Regeneron Pharmaceuticals. D.L. has received research support from Bayer, Biomarin, CSL-Behring, and Sanofi and serves as an advisor for Biomarin, CSL-Behring, Sanofi, and Takeda.
Valder R. Arruda died on 20 May 2022.
Correspondence: Roland W. Herzog, IUPUI-Wells Center for Pediatric Research, 1044 West Walnut St, Indianapolis, IN 46202; e-mail: rwherzog@iu.edu.

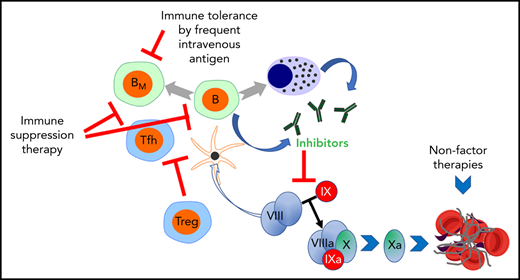
![Clinical immunotherapies and alternative approaches to eliminate or circumvent antibodies against coagulation factors formed in patients with bleeding disorders. These concepts are illustrated using inhibitor formation in the treatment of hemophilia as an example. Alternative strategies include [1] immune tolerance induction (ITI) by frequent IV administration of antigen; [2] transient immune suppression therapy (IST); and [3] restoration of hemostasis using nonfactor therapeutics, thus avoiding the use of the target antigen. Center: Inhibitory antibodies are produced by B cells upon their T helper cell-dependent activation. T follicular helper cells promote the organization of germinal centers and the production of antibodies. B cells may differentiate into memory B cells (BM) or plasma cells (PCs), which produce antibodies long-term. Ultimately, inhibitors prevent blood clot formation by reducing/eliminating the activation of factor X. This reaction is a critical component of the coagulation cascade and normally occurs through the cooperation of activated FVIII (FVIIIa) and factor IX (FIXa). Left: Although little is known about the mechanisms by which ITI reverses inhibitor production upon frequent IV factor administration, evidence supports that high antigen doses can directly inhibit BM. IST may use small molecule drugs such as cyclophosphamide, a DNA alkylating agent that eliminates proliferating cells such as activated B and T cells. Rituximab (anti-CD20 antibody) depletes mature B cells (but not BM or PCs). Regulatory T cells (Tregs) are able to suppress inhibitor formation. Therefore, experimental approaches use inhibition of the mTOR pathway with rapamycin/sirolimus (which aids in deletion of effector T cells, suppresses germinal center formation, and promotes Treg induction); oral antigen administration or hepatic gene transfer to induce Tregs, or Treg cell therapy, among other approaches. Right: Alternatively, nonfactor therapies bypass the effect of inhibitors by using a bispecific antibody that partially mimics the function of FVIII but is not recognized by FVIII inhibitors (a treatment that, however, only applies to HA) or promotes coagulation by elimination/neutralization of critical components of anticoagulant pathways (such as AT or TFPI) through small interfering RNA or monoclonal antibody therapy. AT, antithrombin III; TFPI, tissue factor pathway inhibitor.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/140/10/10.1182_blood.2022016530/3/m_bloodbld2022016530cf1.png?Expires=1771174293&Signature=BZh60DYl3pK9C6aW1BBmDKPmBwvKzWlGZw9R4-xxqXEAWZMRVcWVCbx1xyio8XJBE~Sfw9LPa8XY4~AjlgpeRi1jWEdCxe51xKlvqfK1-IPv7x0Bpn66-8zA~4MawLjbXJ3QRH~MjbN~3w0aP1YCBJx8cUca8GwKsIZDGlMvCX5WbZMxP8hZbSUvNKxT4ZcafOjgdCGeFUPJbr~yontQEBKEjJUOolevQI5Zm4F1vJH2W2RtME70IsWiy~E0RXQCDtltdn1QmCxPpw3-8BtJOM3gyOkMm2evt2Vc2sa-n1cg96QIOz3sq98rzE7VUAvkiaCspYx0-lrT0nJm2FQP8A__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)