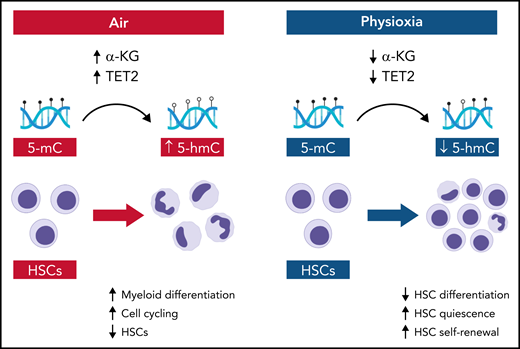
In this issue of Blood, Aljoufi et al1 report that physiologic oxygen tension or “physioxia” downmodulates ten-eleven translocation 2 (Tet2) activity in hematopoietic stem cells (HSCs) and Tet2 regulates HSC differentiation and loss of HSC self-renewal that occurs in response to ambient air.
HSC transplantation provides curative therapy for patients with a variety of hematologic diseases.2 Mantel et al3 previously demonstrated that the usual collection and processing of HSCs in ambient air (∼21% O2) decreases the recovery of HSCs because of extraphysiologic oxygen shock/stress response, which produces increased reactive oxygen species (ROS) and activates mitochondrial permeability transition pores (MPTP). Mantel et al3 further described that collection of murine or human HSCs in physioxia (∼3% O2) substantially increased recovery of HSCs capable of long-term repopulation. This discovery provided the potential for improved methods for human HSC transplantation and human HSC gene editing.4 Aljoufi et al have now elucidated a molecular key that regulates the HSC response to changes in oxygen tension.
In the Aljoufi et al study, the authors performed single-cell RNA-sequencing of HSCs in physioxia and ambient air and determined that expression of the epigenetic modifier Tet2, an α-ketoglutarate (α-KG)-, iron-, and oxygen-dependent dioxygenase that converts 5-methylcytosine (5-mC) to 5-hydroxymethylcytosine (5-hmC),5 was downregulated in HSCs in physioxia (see figure). Importantly, upon exposure to ambient air, wild-type HSCs displayed increased myeloid differentiation and loss of competitive repopulating capacity, whereas Tet2−/− HSCs maintained both multilineage potential and competitive repopulating capacity. Furthermore, physioxia had no significant effect on Tet2−/− HSCs, which maintained long-term competitive repopulating capacity in both ambient air and physioxia. Taken together, these data suggest that physioxia promotes HSC self-renewal, at least in part, through downregulation of Tet2.
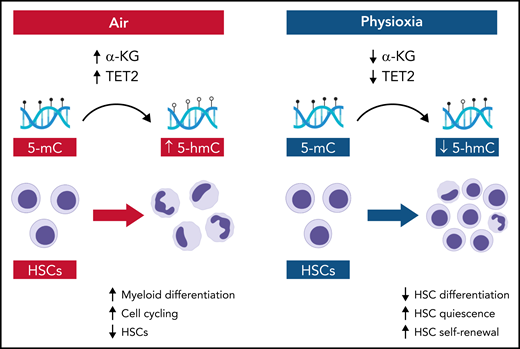
Schematic showing the molecular and cellular effects of ambient air and physioxia on HSCs. In air, α-KG levels and Tet2 activity are relatively increased in HSCs, producing increased conversion of 5-mC to 5-hmC, coupled with HSC differentiation and HSC loss. In physioxia, α-KG levels and Tet2 activity are decreased, which decreases HSC differentiation and increases HSC self-renewal. Illustration by Bryanna C. Reinhardt, Louisiana State University School of Medicine; images/shapes used from BioRender.com.
Schematic showing the molecular and cellular effects of ambient air and physioxia on HSCs. In air, α-KG levels and Tet2 activity are relatively increased in HSCs, producing increased conversion of 5-mC to 5-hmC, coupled with HSC differentiation and HSC loss. In physioxia, α-KG levels and Tet2 activity are decreased, which decreases HSC differentiation and increases HSC self-renewal. Illustration by Bryanna C. Reinhardt, Louisiana State University School of Medicine; images/shapes used from BioRender.com.
Through its role as an epigenetic modifier, Tet2 regulates HSC differentiation to myeloid and erythroid lineages,6 and deletions and loss-of-function mutations in Tet2 occur in 10% to 20% of patients with myelodysplastic diseases and acute myeloid leukemia.7Tet2 mutations are also commonly observed in clonal hematopoiesis of indeterminate potential.8 Animal models have confirmed that loss of Tet2 produces increased HSC self-renewal and expansion of the HSC pool coupled with splenomegaly and extramedullary hematopoiesis, which is consistent with myeloid transformation.7 In their study, Aljoufi et al show that Tet2 also plays an essential role in promoting HSC differentiation in response to oxygen stress. To confirm this point, the authors abrogated the effects of physioxia on wild-type HSCs via supplementation with α-KG, which as a cofactor for Tet2, augmented Tet2 activity, increased conversion of 5-mC to 5-hmC, and increased HSC differentiation.
The results of the Aljoufi et al study raise several interesting questions: first, Does physioxia sustain HSC numbers and HSC self-renewal capacity solely by inhibition of Tet2-mediated oxidation of 5-mC to 5-hmC? Or does physioxia also suppress other important Tet2 epigenetic functions (eg, histone modification via regulation of O-linked β-N-acetylglucosamine transferase)?9 Furthermore, How does the function of Tet2 in regulating the HSC response to oxygen shock/stress relate to the function of cyclophilin D, which Mantel et al3 previously demonstrated to have an integral role in promoting MPTP induction, ROS generation, and HSC loss in ambient air?
The application of physioxia conditions could improve HSC transplantation or HSC gene editing, but it raises practical challenges. The results of the Aljoufi et al study suggest that targeting the molecular mechanisms underpinning physioxia could, in principle, sustain HSC numbers and function ex vivo. Interestingly, Guan et al10 recently described a Tet-specific inhibitor that produced synthetic lethality for Tet2-mutant myeloid neoplastic cells while displaying no pro-proliferative effects on normal hematopoietic progenitor cells. Although even temporary inhibition of the Tet2 tumor suppressor in HSCs would require caution, an alternative strategy could utilize supplementation with 2-hydroxyglutarate, which antagonizes α-KG as a co-factor for Tet2 dioxygenase,6 thereby inhibiting Tet2 enzymatic activity and mimicking physioxia. The article by Aljoufi et al provides an important step toward understanding the molecular mechanisms that govern the HSC response to extraphysiologic stress and how such mechanisms might be targeted.
Conflict-of-interest disclosure: J.C.P. declares no competing financial interests.