

In this issue of Blood, Kaiser et al1 reveal how the reparative process is initiated by ceaselessly vigilant patrolling platelets that turn procoagulant and instigate hemostatic plug formation to seal endothelial gaps in the wake of transmigrating leukocytes.
Platelets as keepers of vascular integrity can prevent hemorrhage in the dense microvascular beds of lungs, gastrointestinal tract, and skin during nonsterile inflammation.2 Whether the molecular mediators of primary hemostasis overlap with or are distinct from those mounting inflammatory hemostasis is an absorbing yet unresolved enigma. Kaiser et al have capitalized on their previous findings, which describe immune-competent migratory platelets3 that sense vascular breaches (see figure).4 An extensive and elegantly performed series of in vitro and in vivo experiments after lipopolysaccharide (LPS)-induced inflammation that affected the mesenteric and pulmonary vasculature shows that migrating platelets on the watch are arrested on collagen to drive inflammatory hemostasis, involving costimulatory cues from glycoprotein IIb/IIIa (GPIIb/IIIa)–mediated outside-in-signaling and GPVI. While providing a translational perspective the investigators have further verified that simultaneous pharmacologic targeting of GPIIb/IIIa and GPVI, also anticoagulants in clinical practice (argatroban, enoxaparin, rivaroxaban) may aggravate alveolar hemorrhage. This is indeed a matter of concern, especially in immunothrombotic diseases such as SARS-CoV-2 (COVID-19), in which lungs are the acute inflammatory hotspots. The procoagulant platelets5 in the circulation of COVID-19 patients provoke a hyper-coagulatory and prothrombotic disposition which worsens prognosis and thus requires therapeutic or prophylactic administration of anticoagulants.
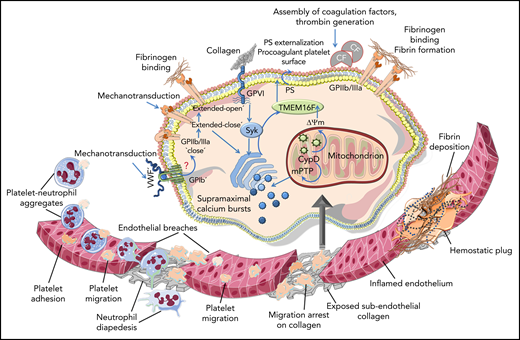
Schematic diagram depicting neutrophils and platelets reaching the inflamed endothelium, where migrating platelets become arrested on the exposed subendothelial collagen at neutrophil extravasation sites, which generates endothelial breaches. Collagen-arrested platelets turn procoagulant upon receiving costimulatory mechanosensing signals from GPIIb/IIIa-Gα13 and GPVI. Syk is activated downstream of GPVI, and supramaximal calcium bursts and CypD-dependent mitochondrial permeability transition pore (mPTP) opening causes mitochondrial depolarization (ΔΨm). This culminates in membrane translocase TMEM16F-mediated PS exposure. PS-assisted assembly of coagulation factors (CFs) on procoagulant platelets drives thrombin generation that results in the formation of fibrin-enriched microthrombi, which restores vascular integrity. However, a potential role of von Willebrand factor (VWF)—GPIb-mediated mechanosensing on the sequential transition of GPIIb/IIIa affinity from a closed to an intermediate extended-closed state essential to mediate mechanotransduction, and eventually to an extended-open state—remains to be deciphered. Illustration was created with the help of Servier Medical art: https://smart.servier.com.
Schematic diagram depicting neutrophils and platelets reaching the inflamed endothelium, where migrating platelets become arrested on the exposed subendothelial collagen at neutrophil extravasation sites, which generates endothelial breaches. Collagen-arrested platelets turn procoagulant upon receiving costimulatory mechanosensing signals from GPIIb/IIIa-Gα13 and GPVI. Syk is activated downstream of GPVI, and supramaximal calcium bursts and CypD-dependent mitochondrial permeability transition pore (mPTP) opening causes mitochondrial depolarization (ΔΨm). This culminates in membrane translocase TMEM16F-mediated PS exposure. PS-assisted assembly of coagulation factors (CFs) on procoagulant platelets drives thrombin generation that results in the formation of fibrin-enriched microthrombi, which restores vascular integrity. However, a potential role of von Willebrand factor (VWF)—GPIb-mediated mechanosensing on the sequential transition of GPIIb/IIIa affinity from a closed to an intermediate extended-closed state essential to mediate mechanotransduction, and eventually to an extended-open state—remains to be deciphered. Illustration was created with the help of Servier Medical art: https://smart.servier.com.
It is well known that platelets seal endothelial breaks that result from leukocyte extravasation to prevent inflammatory hemorrhage because thrombocytopenia aggravates dermal and alveolar bleeding; however, β3-integrin−/− and FcγR−/− mice2 are spared. The study by Kaiser et al revisits the old chapters on this subject to fill in the gaps in our understanding regarding the mechanistic drivers of inflammatory hemostasis. Their investigation delineates the sequence of events whereby costimulatory mechanosignals from GPIIb/IIIa-GPVI6,7 arrest migratory platelets on exposed subendothelial collagen and eventually transform them into a procoagulant ballooning phenotype8 to form a fibrous hemostatic plug, which restores vascular integrity. Upon encountering collagen matrix, but not collagen fibers in solution, migratory platelets become stationary and phosphatidylserine (PS) positive, and they release microvesicles. GPIIb/IIIa is crucial for platelet migration on fibrinogen3,4 to arrive at the exposed collagen hotspots, and it is also crucial for the subsequent procoagulant activity that is abolished upon inhibiting Gα13-mediated outside-in signaling (mP6) without affecting fibrinogen ligation and retaining migratory capacity. Although migrating platelets that circumvent collagen barely adopt a procoagulant phenotype, GPVI-mediated fibrinogen sensing is dispensable for migration on the specified hybrid matrix in vitro. α2β1-Integrin is involved neither in migration nor in PS exposure. However, Syk inhibition downstream of GPVI significantly affects the procoagulant alteration of migratory platelets. The authors have further validated the mechanosensing mode of action6,7 and have demonstrated that procoagulant transformation of migratory platelets occurs independent of biochemical stimuli from soluble agonists. Terutroban (thromboxane receptor), cangrelor (P2Y12), vorapaxar (PAR1) and BMS-986120 (PAR4) do not interfere with platelet migration or procoagulant activity on the hybrid matrix in vitro, suggesting a differential impact on pathological thrombosis without compromising inflammatory hemostasis.
Costimulation of GPIIb/IIIa-transduced outside-in signaling and GPVI triggers supramaximal calcium mobilization on collagen-arrested platelets and causes cyclophilin D (CypD)–dependent mitochondrial depolarization (ΔΨm), which culminates in membrane translocase TMEM16F-mediated PS exposure. PS-assisted assembly of coagulation factors (CFs) and thrombin generation results in the formation of fibrin-enriched microthrombi, which reinstates vascular integrity. Consequently, although migration, degranulation, and GPIIb/IIIa activation remain unaffected, genetic ablation or pharmacologic inhibition of CypD or TMEM16F compromises procoagulant activity. This exaggerates inflammation-induced alveolar hemorrhage without the compensatory restoration offered by procoagulant platelets at transendothelial extravasation sites, because neutrophil infiltration remains unchanged. Tirofiban, which prevents GPIIb/IIIa fibrinogen binding, reduces PS exposure on migratory platelets, an effect that is further reinforced by GPVI blocking or depletion (JAQ1). Therefore, concurrent GPIIb/IIIa-GPVI antagonism aggravates alveolar hemorrhage and reduces the proportion of procoagulant platelets in circulation, which causes mesenteric microbleeding after LPS challenge. How mechanotransduction through GPIIb/IIIa-GPVI is influenced by biomechanical determinants that drive variations in blood hemodynamics across diverse vasculatures in vivo remains a matter of speculation. Differences in endothelial phenotypes and their response to specific infectious or inflammatory cues across varied vascular beds may further add to the complexity of crosstalk between endothelium, platelets, and the immune, coagulation, and fibrinolytic systems. Nevertheless, Kaiser et al convincingly demonstrate the mechanistic mediation of GPIIb/IIIa-GPVI in LPS-induced inflammatory hemostasis in the lungs and postcapillary mesenteric venules. However, the impact of von Willebrand factor and GPIb-mediated mechanosensitivity on the sequential transition of GPIIb/IIIa affinity from a “closed” to an intermediate “extended-closed” state (required to receive mechanosignals and undergo mechanical affinity maturation) to an “extended-open” state6 remains to be elucidated. Moreover, platelet mechanosensing on collagen matrices increases with substrate stiffness, prompting PS exposure.7 How and whether this mirrors the optimal trigger for restorative or deleterious inflammatory hemostasis in a hypercoagulatory vascular microenvironment remains undefined. Although the thromboinflammatory CLEC-2–podoplanin axis9 is suggested to have a minor role in primary hemostasis, it deserves attention, particularly considering the prevalence of podoplanin at sites of vascular breaches, its protective role in attenuating acute lung injury,9 and the involvement of Syk, shared by GPVI and CLEC-2 signaling9 in the procoagulant transformation of migratory platelets.
Curiously, platelets help leukocytes accumulate at inflammatory loci, whereas a platelet-assisted hemostatic plug seals endothelial gaps left by transmigrating leukocytes. Inflammatory hemostasis may be substantiated but not entirely reliant on circulatory platelet-leukocyte interactions, unlike the influence thromboinflammatory platelet-leukocyte aggregates have in atheroprogression, in venous thrombosis, or in influencing thrombotic propensity after ischemia reperfusion.10 Therefore, simultaneous GPIIb/IIIa-GPVI blockade does not reduce platelet-neutrophil aggregates or pulmonary recruitment of neutrophils after LPS challenge, but it does exacerbate alveolar microbleeds that negatively correlate with procoagulant platelet counts. Accordingly, anticoagulants aggravate pulmonary hemorrhage without affecting circulatory platelet-neutrophil aggregates or pulmonary neutrophil infiltration. Nonetheless, neutrophil depletion in LPS-challenged mice affects intravascular fibrin(ogen) deposition in mesenteric vessels, suggesting that neutrophils and platelets may cooperate to reach the inflamed endothelium, but the crucial procoagulant transformation of platelets requires their arrest on collagen and the mechanosensing services of GPIIb/IIIa-GPVI. The therapeutic goal is to sustain hemostatic plugging of the inflamed vessel and prevent pathological thrombosis at the same time. This requires further insights into the intricate details of classical and inflammatory hemostasis. Potential drug targets may emerge from subtle differences between PS-exposing apoptotic vs procoagulant platelets, PAC-1–binding vs PS+ platelets, and the mechanosensing vs signaling and adhesive involvement of GPIIb/IIIa and GPVI. Therefore, we can expect truly translational findings from continued investigation on the intriguing platelet patrol.
Conflict-of-interest disclosure: The author declares no competing financial interests.

