In this issue of Blood, Zhang et al1 reported that extracellular signal-regulated kinase 2 (ERK2) substrate-binding domains have opposing roles in Janus kinase 2 V617F (JAK2V617F)-driven myeloproliferative neoplasms (MPNs). One of the domains, ERK2-docking (ERK2-D), may be a promising therapeutic target for MPNs. Conversely, the other domain, ERK2-DEF–binding pocket (ERK2-DBP), blocks progression of the disease in a mouse model. This changes the perception that simply inhibiting the catalytic activity of ERK1/2 in MPNs would be an effective therapeutic strategy.
MPNs are chronic disorders, and patients present with symptoms that affect their quality of life and have a significant risk of thrombotic complications, fibrotic evolution, and leukemic transformation.2 There is a real need to improve the understanding of MPN biology and develop new combinations of targeted therapies to eradicate the MPN clone by shutting off all the activated pathways in the MPN cells.
MPNs are hematopoietic stem cell diseases characterized by clonal expansion of 1 or more myeloid lineages resulting in erythrocytosis (polycythemia vera [PV]), in thrombocytosis (essential thrombocythemia [ET]), or in progressive fibrosis in the bone marrow (primary and post PV/ET myelofibrosis [MF]). All these chronic states of disease can evolve into highly aggressive acute myeloid leukemia with poor survival. MPNs are driven by a mutation of 1 of the 3 driver genes—Janus kinase 2 (JAK2), calreticulin (CALR), and myeloproliferative leukemia virus (MPL).2 The resulting constitutive activation of JAK2 signaling activates STAT, PI3K/AKT, and MEK/ERK signaling.2
JAK2 inhibitors are now used for the treatment of MF and PV with clinical benefits such as a decrease in inflammatory symptoms and splenomegaly. However, unlike imatinib and other kinase inhibitors in BCR-ABL–driven chronic myeloid leukemia, JAK2 inhibitors do not significantly decrease the MPN clone, and MPN cells can survive and proliferate when treated with JAK2 inhibitor monotherapy.3 Several experimental approaches have shown that activation of mitogen-activated protein kinase (MAPK) pathway signaling (MEK/ERK) contributes to the JAK2 inhibitor persistence or resistance in MPNs.4,5
ERK1/2 are terminal serine/threonine kinases of the MAPK signaling pathway that act on multiple downstream targets involved in cellular proliferation and survival. In mouse models of MPNs, ERK1/2 deficiency reduced the frequency of JAK2V617F clones and decreased the features of MPNs.6 Combining the JAK2 inhibitor ruxolitinib with ERK inhibitors normalized erythrocytosis and splenomegaly in mice with JAK2V617F MPNs with an interesting long-term clone reduction.6
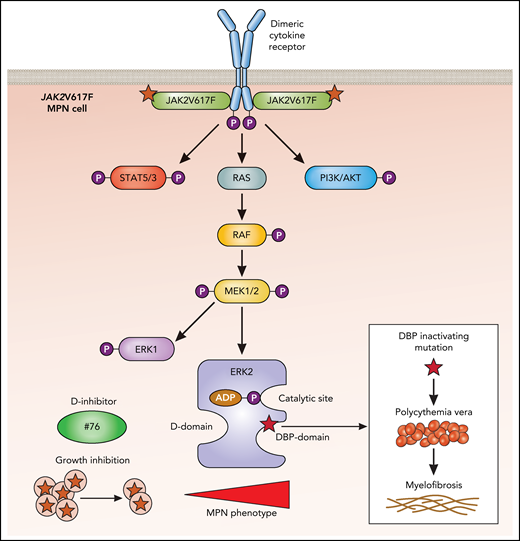
ERK2 interacts with substrates through 2 domains on opposing faces of the protein (see figure). The D domain binds Leu-X-Leu or hydrophobic sequences proximal to basic residues,7 and the DBP domain (docking site for ERK and FXF [DEF]–DBP) binds sequences that exhibit Phe-X-Phe followed by a Pro residue.8
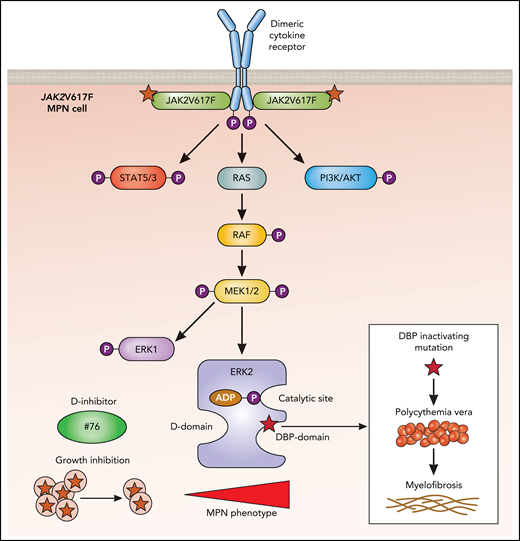
In MPNs, dimeric cytokine receptors such as erythropoietin and thrombopoietin receptors are bound by the active JAK2V617F mutant, which endows the receptor with the ability to signal persistently in the absence of or with very low levels of ligands. This results in persistent activation of the indicated downstream pathways, which includes ERK2 (blue). Shown are the 2 substrate-binding domains: the D domain and the DBP domain. Although a small molecule inhibitor (compound #76) blocking the D domain reduces growth of JAK2V617F cells and reduces the MPN phenotype, the inactivation of the DBP domain leads to an enhancement of the MPN phenotype, with progression of PV to MF. ATP, adenosine triphosphate; P, phosphorylation. Professional illustration by Patrick Lane, ScEYEnce Studios.
In MPNs, dimeric cytokine receptors such as erythropoietin and thrombopoietin receptors are bound by the active JAK2V617F mutant, which endows the receptor with the ability to signal persistently in the absence of or with very low levels of ligands. This results in persistent activation of the indicated downstream pathways, which includes ERK2 (blue). Shown are the 2 substrate-binding domains: the D domain and the DBP domain. Although a small molecule inhibitor (compound #76) blocking the D domain reduces growth of JAK2V617F cells and reduces the MPN phenotype, the inactivation of the DBP domain leads to an enhancement of the MPN phenotype, with progression of PV to MF. ATP, adenosine triphosphate; P, phosphorylation. Professional illustration by Patrick Lane, ScEYEnce Studios.
To understand the roles of ERK2 substrate-binding domains in MPN pathogenesis, Zhang et al generated an ERK2-mutant knockin mouse model with an inactivated ERK2-DBP domain but with preserved ERK2 kinase activity and D domain function. Wild-type (WT) mice developed an expansion of the immature erythroid compartment with splenomegaly. JAK2V617F was transduced into the bone marrow stem and progenitor cells that expressed ERK2-DBP mutant, ERK2-WT, or ERK2 knockout. When transduced cells were transplanted into sublethally irradiated immunodeficient mice, ERK2-DBP–mutant recipients developed PV and rapidly progressed to MF at 12 weeks.
The mechanism that explained this unexpected effect, which involved the 3 myeloid lineages instead of the predicted lineage was explored in vitro. Although ectopic JAK2V617F expression in bone marrow stem and progenitor cells reduced colony formation in WT mouse cells via inducing oncogene-induced senescence, this was not the case for JAK2V617F cells expressing the ERK2-DBP mutant. Thus, the ERK2 DBP mutant seemed to reduce senescence in vitro. Mechanistically, an inactive DBP domain would not allow physical interaction of ERK2 with Egr-1, a transcription factor that contains a DEF domain binding to the ERK2-DBP domain, and which was required for the induction of senescence by JAK2V617F. These in vitro results need to be confirmed in vivo.
Given that ERK2-DBP domain antagonized the progression of JAK2V617F-driven MPNs, the authors hypothesized that the remaining ERK2-D domain must play a major role in promoting disease progression. To explore this hypothesis, Zhang et al developed a selective inhibitor (compound #76) of the ERK2-D domain. Encouragingly, compound #76 reduced in vivo growth of injected tumoral JAK2V617F-expressing SET2 cells in immunodeficient mice. Again, these data need confirmation in a bona fide mouse model of MPNs.
The key question raised by this study is the nature of the ERK2-D domain substrates contributing to JAK2V617F-driven MPN disease and progression. Do these findings apply to MPN cells driven by driver mutations other than JAK2V617F? For example, in mouse models expressing the active MPL W515A/K, ERK1/2 signaling promoted the MPN phenotype and MF.9 Finally, certain ERK2 substrates contain both D and DEF domains (such as Elk1), and the 2 substrate-binding sites communicate spatially in ERK2.10 It would be of interest to assess whether the substrates that contain both substrate domains contribute to the phenotype of MPNs.
JAK2V617F-driven MPNs remain incurable diseases. Selectively targeting the D domain of ERK2 and possibly key substrates binding to the D domain may become promising avenues of investigation for novel therapies.
Conflict-of-interest disclosure: The authors declare no competing financial interests.