Key Points
Compared with tisa-cel, axi-cel was associated with better disease control but had a less favorable safety profile in SOC treatment of LBCL.
Other outcome determinants included bridging success, performance status, age, LDH, prolonged neutropenia, and/or severe neurotoxicity.

Abstract
CD19-directed chimeric antigen receptor (CAR) T cells have evolved as a new standard-of-care (SOC) treatment in patients with relapsed/refractory (r/r) large B-cell lymphoma (LBCL). Here, we report the first German real-world data on SOC CAR T-cell therapies with the aim to explore risk factors associated with outcomes. Patients who received SOC axicabtagene ciloleucel (axi-cel) or tisagenlecleucel (tisa-cel) for LBCL and were registered with the German Registry for Stem Cell Transplantation (DRST) were eligible. The main outcomes analyzed were toxicities, response, overall survival (OS), and progression-free survival (PFS). We report 356 patients who received axi-cel (n = 173) or tisa-cel (n = 183) between November 2018 and April 2021 at 21 German centers. Whereas the axi-cel and tisa-cel cohorts were comparable for age, sex, lactate dehydrogenase (LDH), international prognostic index (IPI), and pretreatment, the tisa-cel group comprised significantly more patients with poor performance status, ineligibility for ZUMA-1, and the need for bridging, respectively. With a median follow-up of 11 months, Kaplan-Meier estimates of OS, PFS, and nonrelapse mortality (NRM) 12 months after dosing were 52%, 30%, and 6%, respectively. While NRM was largely driven by infections subsequent to prolonged neutropenia and/or severe neurotoxicity and significantly higher with axi-cel, significant risk factors for PFS on the multivariate analysis included bridging failure, elevated LDH, age, and tisa-cel use. In conclusion, this study suggests that important outcome determinants of CD19-directed CAR T-cell treatment of LBCL in the real-world setting are bridging success, CAR-T product selection, LDH, and the absence of prolonged neutropenia and/or severe neurotoxicity. These findings may have implications for designing risk-adapted CAR T-cell therapy strategies.
Introduction
Patients with large B-cell lymphoma (LBCL), relapsed/refractory (r/r) after multiple lines of therapy, or autologous stem cell transplantation generally have a poor prognosis with a median overall survival (OS) of only a few months.1-4 Since August 2018, 2 CD19-targeting chimeric antigen receptor (CAR) T-cell constructs, tisagenlecleucel (Kymriah, tisa-cel, Novartis) and axicabtagene ciloleucel (Yescarta, axi-cel, Gilead) are commercially available for the treatment of r/r LBCL in Europe. The pivotal clinical trials leading to approval of these CAR T-cell constructs have demonstrated an excellent rate of overall response (ORR) and complete remission (CR) of 52% and 40% with tisa-cel, and 82% and 54% with axi-cel, respectively.5,6 Approvals were based on relatively small phase 2 studies, including up to 111 patients due to high medical need in this heavily pretreated patient population, with only limited alternative curative treatment options. Therefore, data on patient selection, toxicity, early and late efficacy, and long-term outcome using these new but costly therapeutic tools in a standard-of-care (SOC) setting are urgently required.
Unanswered questions of particular importance relate to the safety of CD19 CAR T-cell therapies when used outside of clinical trials, predictors of outcome, and the impact of bridging strategies to control disease before CAR T-cell treatment. Furthermore, possible differences in the safety and efficacy profiles of the 2 products currently available remain to be determined in a real-world setting. To address these questions, we took advantage of registry data collected by the German Registry for Stem Cell Transplantation (Deutsches Register für Stammzelltransplantation, DRST) as per federal German regulations.
Patients and methods
Data source
The DRST is a voluntary organization of all German adult transplant centers. Of 26 centers performing CAR T-cell therapy at the time of this analysis, 21 participated in this study. The DRST performs data collection of cellular therapies in cooperation with the European Society of Blood and Marrow Transplantation (EBMT) using the EBMT ProMISe (Project Manager Internet Server) database. Accreditation as a DRST center requires submission of minimal essential data (EBMT MED-A form) from all consecutive cellular therapy recipients to the EBMT central database in which patients can be identified by the diagnosis of underlying disease and type of cellular therapy. EBMT/DRST registry data are routinely audited to determine the accuracy of data collected as part of the Joint Accreditation Committee International Society for Cell and Gene Therapy–Europe and EBMT certification. Data collection requires written informed consent using a consent form based on a standard DRST/EBMT template following the European data protection regulations and the Declaration of Helsinki.
Study design
This study was performed under the auspices of the Working Group Hematopoietic Cell Therapy of the German Lymphoma Alliance (GLA), which is the German National Lymphoma Study Group. Participating GLA member centers are committed to sharing their data for this project. All centers were trained and qualified by the respective manufacturers for CAR T-cell application and management of toxicities. Furthermore, as per federal directive, all centers were obliged to adhere to defined quality assurance measures based on the guidelines of the German Board of Oncology/Hematology for the management of CAR T-cell toxicities (www.onkopedia.com/de/onkopedia/guidelines).
Data were approved and provided for this study by the DRST. Baseline patient, disease, cellular therapy, and outcome data were obtained from registry files (MED-A cellular therapy forms). Centers were contacted to provide additional treatment and follow-up information using additional data fields. Collected data underwent further quality control, source data review, and additional validation by the study office in Tuebingen, Germany, and the local investigators. The study was performed in accordance with the Declaration of Helsinki and approved by the Ethical Committee of the University of Tuebingen (Ref. Nr. 277/2020BO2).
Those eligible were adult (age ≥18 years) patients with LBCL treated and dosed with commercially available tisa-cel or axi-cel and documented in the EBMT/DRST database from November 2018 through April 2021. Patients treated with other CAR T-cell constructs or within clinical trials were excluded.
The objective of the study was to assess the efficacy and toxicity of commercial CAR T-cell therapy for LBCL in a real-world setting in Germany. The following outcome parameters were analyzed: ORR, CR, OS, progression-free survival (PFS) rate, nonrelapse mortality (NRM), incidence and severity of cytokine release syndrome (CRS), and immune effector cell-associated neurotoxicity syndrome (ICANS). CRS and ICANS were graded according to the American Society for Transplantation and Cellular Therapy grading criteria.7 Bridging therapy was defined as any therapy received for lymphoma control between leukapheresis and the start of lymphodepleting chemotherapy. Neutrophil or platelet recovery was defined as achievement of absolute neutrophil count >500/µL or platelet count >20 000/µL.
Statistical analyses
Descriptive statistics used absolute frequencies and percentages for categorical variables and medians and ranges for continuous variables. Differences between groups were assessed using χ-square tests and Mann-Whitney rank-sum tests. Probabilities of OS and PFS were estimated using Kaplan-Meier plots and log-rank tests to identify differences between groups. Cumulative incidence was used to estimate NRM and relapse with relapse/progression and death of any cause as a competing event. OS was defined as the time from cellular therapy to death from any cause, and PFS was defined as the time from cellular therapy to relapse or disease progression or death from any cause, whatever came first. NRM was defined as death after cellular therapy without prior lymphoma relapse or progression. Simple and multiple Cox regression analysis was applied to further investigate predictive factors for OS and PFS. In multiple Cox regression, forward variable selection with inclusion/exclusion probabilities 0.05/0.10 was applied. Results are expressed as hazard ratio (HR) with a 95% confidence interval (CI). All tests and CIs were 2-sided. The level of significance was 0.05 for all tests. Analyses were performed by SPSS 26.0 (SPSS Inc., Chicago, IL) and GraphPad Prism Software 9.1.2 (GraphPad Software, La Jolla, CA). Incidence curves taking into account competing risks were analyzed using R software (R Foundation for Statistical Computing, Vienna, Austria) cmprsk package.8
Results
Patient characteristics
A total of 356 consecutive patients treated in 21 German CAR T-cell centers (of 26 performing CAR T-cell therapy in the index period) were included; 183 patients were treated with tisa-cel and 173 patients with axi-cel. Nine of 21 centers treated their patients with both constructs, 7 of 21 only with tisa-cel, 5 of 21 only with axi-cel. Baseline characteristics were well balanced between axi-cel and tisa-cel recipients except for a significantly higher proportion of patients with bridging treatments and being refractory at the start of lymphodepletion in the tisa-cel group (Table 1). The median age was 60 years (range, 19-83), and 66% of patients were male. About half of the patients (52%) had a high/high-intermediate international prognostic index (IPI)9 at the start of lymphodepleting chemotherapy. Furthermore, the majority of patients (60%) had an elevated lactate dehydrogenase level (LDH) before lymphodepletion. Performance status was Eastern Cooperative Oncology Group (ECOG) ≥2 in 16% of the patients. The majority (n = 252 [71%]) had received ≥3 pretreatment lines before the start of bridging therapy or lymphodepletion. Of these, 20% (n = 51/252) had received ≥5 pretreatment lines. Prior autologous (n = 108) or allogeneic (n = 13) hematopoietic cell transplantation (HCT) had been performed in 34% of the patients; 76% (n = 269/356) of the patients were defined as refractory to chemotherapy before the start of lymphodepletion by the treating physician. Patients were evaluated for potential eligibility for either the ZUMA-1 or the JULIET trial, retrospectively (Table 1).
Bridging therapy for disease control before lymphodepletion was administered in 78% (n = 278) of the patients. The patients receiving bridging therapy were associated with higher IPI and elevated LDH: IPI ≥3, no bridging: 28/76 (37%) vs bridging: 154/269 (57%); P = .0018; or LHD > ULN, no bridging: 34/76 (45%) vs bridging: 172/269 (64%); P = .0034. As was no response to bridging: IPI ≥3 response: 23/59 (38%) vs 131/210 (63%); P = .0017; no response or LDH > ULN response: 30/59 (51%) vs 142/210 (68%); P = .022; no response (supplemental Table 4). Bridging modalities included classical platinum-based chemoimmunotherapy (n = 67, rituximab/ifosfamide/carboplatin/etoposide [R-ICE] = 27, R-gemcitabine/oxaliplatin = 33, other = 7) or regimens of similar intensity (n = 71 [26%]), polatuzumab-based regimens (n = 71 [26%]), other rituximab-based chemoimmunotherapy (n = 33 [12%]), radiotherapy (n = 30 [11%]), immunotherapy (n = 12 [4%]), and steroids (n = 6 [2%]) (supplemental Table 1). Of the 71 patients receiving polatuzumab as first bridging therapy, 52 also received bendamustine. Overall, 57 patients were exposed to bendamustine in the bridging regimen, 38 in the tisa-cel, and 19 in the axi-cel cohort. Bridging resulted in disease control (CR/partial remission [PR]) in 59 of 269 patients (22%) evaluable for response (supplemental Table 4), with polatuzumab-based regimens tending to result in superior response rates (34%) (supplemental Table 1).
The median time from the tumor board’s decision recommending CAR T-cell therapy to CAR T-cell infusion was 68 days (range, 28-278; axi-cel = 65, tisa-cel = 68). The median time from apheresis to CAR T-cell infusion was 42 days (range, 27-526; axi-cel = 35, tisa-cel = 55).
Toxicity
Grade 4 neutropenia (<500 neutrophils/µL) was reported in 261 of 319 (81%) patients evaluable. The median duration of grade 4 neutropenia was 13 days (range, 1-419). Of 312 patients with PFS ≥28 days, 53 (17%) had persisting grade 4 neutropenia at that time point. The cumulative incidence of neutrophil recovery at day 28 and day 100 was 74% and 90%, respectively. Severe thrombocytopenia (<20 000 platelets/µL) was observed in 115 of 311 (37%) patients evaluable. The median duration of severe thrombocytopenia was 34 days (range, 2-375). In 37/311 (12%) patients, platelet recovery did not occur until the end of follow-up. Cumulative rates of platelet recovery at day 28 and day 100 were 33% and 68%, respectively. There was no difference between the CAR T-cell constructs with respect to the occurrence and extent of cytopenia (supplemental Table 2).
Any grade CRS was observed in 73% of patients with a significantly higher incidence in the axi-cel group (81% vs 65%; P = .0003) (Table 2). CRS grade ≥3 was observed in 12% of patients. ICANS of any grade was observed in 33% of the patients, also with a significantly higher incidence in the axi-cel group (44% vs 22%; P < .0001). ICANS grade ≥3 was reported in 11% of patients, again significantly more common in the axi-cel cohort (P = .004). The median hospitalization time after CAR T-cell therapy was 21 days (range, 8-128).
NRM, relapse, and causes of death
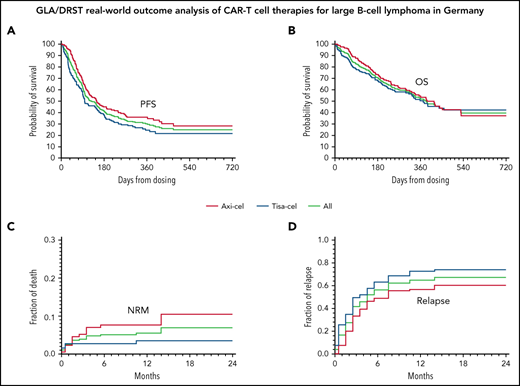
Overall, 164 patients (46%) died, 143 (40%) subsequent to disease progression, and 21 (6%) for nonrelapse-related reasons. Cumulative incidence of NRM adjusted for competing risk of relapse at 12 months was 5.5%; the difference in the incidence of NRM between axi-cel and tisa-cel at 24 months was 10.4% vs 3.5% (P = .032) (Figure 1C). Cumulative incidence of relapse at 12 months adjusted for competing risk NRM was 67%; at 24 months, 74% with tisa-cel and 60% with axi-cel (P = .0004) (Figure 1D). Causes of nonrelapse death were infections (n = 13 [62%]; bacterial, n = 8; fungal, n = 3; viral including 1 case of SARS-CoV2, n = 2); unspecified, n = 1; neurotoxicity (n = 2, 10%), CRS, bleeding, hyperinflammatory syndrome, unknown, and secondary neoplasia (each in 1 patient). Although neurotoxicity was not a major direct reason of death, grade ≥3 neurotoxicity preceded bacterial/fungal infection-related death in 7 of 11 (64%) cases. A detailed description of the patients succumbing to NRM is presented in supplemental Table 3.
Survival, NRM, and relapse. PFS (A) and OS (B) of all (green curves), axi-cel (red curves), and tisa-cel (blue curves), respectively. Cumulative incidence of NRM (C) and relapse (D) adjusted for competing risk.
Survival, NRM, and relapse. PFS (A) and OS (B) of all (green curves), axi-cel (red curves), and tisa-cel (blue curves), respectively. Cumulative incidence of NRM (C) and relapse (D) adjusted for competing risk.
Of note, 14 (67%) NRM events occurred beyond day +28, with infections (n = 9; bacterial = 5, fungal = 2, viral = 2) being the leading direct cause of death. When comparing the characteristics of the 14 patients who experienced “late” NRM with those of the 298 patients who survived day +28 without progression and did not have subsequent NRM, we did not find significant differences in terms of age, sex, IPI, ECOG, pretreatment lines, prior HCT, disease status at lymphodepletion, and prior grade ≥3 CRS. However, a significantly larger proportion of patients with late NRM had persistent grade 4 neutropenia at day +100 or last follow-up (29% vs 5%; P = .0083), had experienced grade ≥3 ICANS (43% vs 9%; P = .002), and/or had received axi-cel (93% vs 51%; P = .0017). Patients with neutrophil nonrecovery and/or grade ≥3 ICANS had a 12-month late NRM incidence of 16% (95% CI, 5.1-26.9) vs 2.5% (95% CI, 0.3-4.7) in patients with none of these 2 factors (supplemental Figure 1).
Outcome
The best responses were CR in 126 (37%) patients, PR in 96 (28%) patients, and stable disease in 38 (11%) out of 344 patients evaluable for response, corresponding to an ORR of 65% (222/344). The median time-to-response was 43 days (range, 5-459). PD was observed in 85 (24%) patients. ORR (CR) in the axi-cel cohort was 74% (42%) compared with 53% (32%) in the tisa-cel cohort; P < .001. With a median follow-up of patients alive of 11 months (range, 1-29), Kaplan–Meier-estimated PFS and OS rates at 12 months were 30% and 52%, respectively (Figure 2). Depending on the CAR-T construct used, axi-cel or tisa-cel, we observed a better PFS with 35% vs 24% at 12-months (P = .015, Log-rank test) but no significant difference in OS (55% vs 53%) (Figure 1A-B).
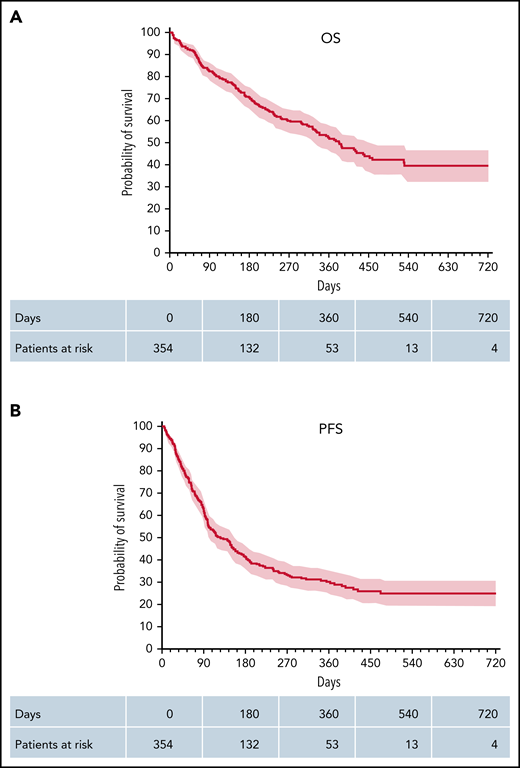
OS and PFS. Kaplan-Meier plots of OS (A) and PFS (B) of all patients. Kaplan-Meier estimated OS and PFS, including 95% CIs (shaded red).
OS and PFS. Kaplan-Meier plots of OS (A) and PFS (B) of all patients. Kaplan-Meier estimated OS and PFS, including 95% CIs (shaded red).
On univariate analysis, significant predictors of an adverse PFS were IPI >2 (P = .004), elevated LDH (P = .002), ineligibility for ZUMA-I (P = .026), nonresponse to bridging therapy (P < .001), and use of tisa-cel (P = .009, Cox model, Wald test). On the contrary, age, performance status (PS), time from board decision to CAR T-cell infusion, and ≥3 lines of preceding therapies had no significant impact. Twelve-month PFS rates for patients without bridging, successful bridging, and bridging failure were 41%, 53%, and 20%, respectively (P ≤ .001). Likewise, increased LDH at lymphodepletion had a negative impact on PFS (Figure 3). Whereas increased LDH at lymphodepletion was not significantly associated with PFS in the group of bridging responders (HR, 0.69; P = .36), it affected the outcome of those patients who did not respond or did not undergo bridging (HR, 1.70; P < .001). The interaction between successful bridging and increased LDH at lymphodepletion was significant (P = .031).
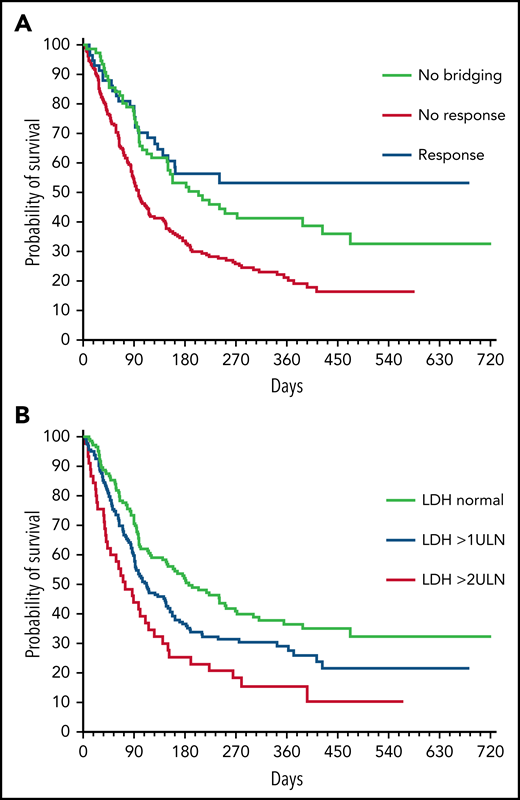
PFS in subgroups. PFS according to (A) bridging response and (B) LDH levels at lymphodepletion.
PFS in subgroups. PFS according to (A) bridging response and (B) LDH levels at lymphodepletion.
Significant predictors of inferior OS on univariate analysis were IPI Score >2 (P = .002), ECOG score >1 (P < .001), elevated LDH at lymphodepletion (P < .001), ineligibility for ZUMA-1 (P = .010), and nonresponse to bridging therapy (P = .001). Three or more lines of preceding therapies and tisa-cel were not associated with inferior OS on univariate statistics.
On multivariate analysis (considering age, IPI, ZUMA-1 eligibility, pretreatment lines, bridging, LDH, PS, and construct), nonresponse to bridging, elevated LDH, and poor performance status were associated with both significantly inferior PFS and OS. In addition, tisa-cel and increasing age were independent risk factors for PFS but did not remain in the final model for OS, while PS significantly affected OS but not PFS (Table 3).
PFS and OS of the 13 patients with prior allogeneic HCT (alloHCT) were not different from that of alloHCT-naïve patients (supplemental Figure 3). Three nonrelapse deaths occurred, 1 each from CRS-related bleeding, late sepsis, and delayed neurotoxicity10 (supplemental Table 3). graft-versus-host disease-associated complications were not reported.
Discussion
The introduction of CAR T cells in the therapeutic armamentarium for the treatment of aggressive B-cell lymphoma marked a paradigm change in lymphoma therapy. Approval studies fostered great hopes with ORR of 54% to 82% and CR rates of 40% to 54%.5,6 Long-term remissions enduring >9 years have been reported.11 However, several important questions remain, including the feasibility and outcomes of this new therapeutic tool in real-world settings and factors determining adequate patient selection for CAR T-cell therapy. Up to now, various groups have reported real-world data in the United States and Europe.12-19 In Germany, CAR T cells have been available for clinical application since the fall of 2018, and as of January 2020, 26 centers had been qualified for CAR T-cell therapy.
The main aims of this study were (1) to investigate the safety of CD19 CAR T-cell therapies when used in a SOC setting, (2) to identify predictors of outcome with a specific focus on the impact of bridging strategies, and (3) to detect possible differences in the safety and efficacy profiles of the 2 products currently available, considering the particularities of the health care system, organizational factors, and patient selection practice effective in Germany.
The incidence and severity of CRS and ICANS in our study were similar as compared to the approval trials, including more neurotoxicity in the axi-cel group. The same accounts for neutropenia, where about 25% and 10% of our patients had not achieved neutrophil recovery by day +28 and day +100, respectively. This is in line with cytopenia patterns reported in the approval trials,6,20 but markedly inferior to hematopoietic recovery kinetics observed after alloHCT for LBCL.21 However, there has been only scant information on if and how neurotoxicity and delayed neutrophil recovery may translate into NRM in the SOC setting. To this end, our data clearly suggest that higher grade neurotoxicity and prolonged neutropenia are associated with an increased risk of subsequent fatal infections, which may occur as late as >1 year after dosing. Late infections after CD19-directed CAR T cells, including severe and fatal courses, have been observed in several small single-center case series,22-25 and the axi-cel real-world milestone analyses from the US, in which infections were a major contributor to NRM.13,17 However, the present study, for the first time, addresses late infectious fatalities and their relation to preceding CAR T-cell–specific complications on a relatively large sample in a multicenter SOC setting in depth. While the relation between neutropenia and infection is obvious, neurotoxicity may predispose to infectious complications through the use of high-dose steroids or alternative immunosuppressants. This stresses the need for close follow-up, immunoglobulin level determination, infectious disease surveillance, and prophylaxis, especially in patients after CAR T-cell therapy being at high risk for developing these complications.26-29
While OR and CR rates with 74% and 42% and 53% and 32% for axi-cel and tisa-cel, respectively, and also OS rates roughly comparable to previously reported experience,5,6 PFS tended to be lower with 12-month rates of 35% and 24% for axi-cel and tisa-cel, respectively. However, our SOC patient cohort differed in several aspects from the patient populations reported in the clinical trials as more patients with reduced performance status and need for bridging were included. Bridging was not allowed in the ZUMA-1 study but used in 78% of patients in our cohort, explaining why only 13% of our patients would have been eligible for inclusion in the ZUMA-1 trial. ZUMA-1 eligibility criteria were explored as a risk factor across the whole sample for allowing a homogeneous analysis, but also because they are more restrictive than the JULIET criteria and considered critical for general CAR T-cell eligibility checks by European cost payers. If JULIET criteria were applied only to tisa-cel patients and ZUMA-1 criteria only to axi-cel patients, trial eligibility did not have a significant impact on outcome (supplemental Figure 3).
The median time from board decision to CAR T-cell infusion was 68 days and thus rather long, reflecting administrative and health insurance clearance hurdles in Germany. Of note, the interval length between indication and dosing was similar for both products and had no impact on PFS. In contrast, the interval between apheresis and CAR T-cell infusion was significantly longer in the tisa-cel cohort, most likely due to the option of cryopreserving the apheresis product before insurance coverage clearance and the start of bridging therapy. Notably, a similar vein-to-vein time was reported for tisa-cel in the BELINDA trial.30
Bridging and its effectiveness was a special focus of our analysis. While the need for bridging before the start of lymphodepletion has been previously described as a risk factor,14,31 we show that bridging success can predict CAR T-cell therapy outcome, thereby supporting the rationale for bridging strategies. One may argue that bridging response might be a surrogate for good risk rather than a therapeutic goal, per se. This view is supported by the fact that bridging nonresponders were characterized by a higher proportion of patients with elevated LDH and advanced IPI. However, this question could only be definitively addressed by a randomized trial. A wide variety of decision strategies for the application of bridging therapy and choices of bridging modalities ranging from steroid monotherapy to high-dose alkylators with autologous stem cell rescue have been used in our patient sample, with polatuzumab-based regimens yielding the most promising results. However, even with polatuzumab, only a minority could be bridged successfully. To this end, novel targeted agents currently entering the clinical stage may help to make CAR T-cell treatment in LBCL more effective in the near future.32-35 Apart from bridging, other factors predicting unfavorable outcomes in our analysis were LDH elevation at lymphodepletion and poor performance status, in line with previous reports.36,37
In contrast to previously published real-world analyses of CD19-directed CAR T-cell therapies, for the first time, we have attempted to compare the safety and efficacy profiles of the 2 constructs available in Europe. The results suggest that when compared with tisa-cel, axi-cel was associated with higher complication rates and higher efficacy but not higher OS. Given the increasing armory of effective salvage therapies for LBCL, the lack of OS difference may be related to the rather short median follow-up of 11 months. Similar results have been recently reported in meeting abstracts by Bachy and colleagues for the French CAR T-cell registry.38 Furthermore, randomized phase 3 studies of the role of either axi-cel or tisa-cel as second-line therapy in comparison with the standard of care only showed positive results for axi-cel.30,39 Axi-cel tended to be associated with higher NRM, partly driven by the complications of neurotoxicity and cytopenia. However, despite quite vigorous safety measures mandated by the authorities and effective national guidelines, risk management practices could vary from center to center and may have influenced results relating to differences between the 2 products and the incidence of late toxicities. Despite this caveat, axi-cel appears to provide superior PFS because of higher response rates and fewer progression events. However, some obvious risk imbalances between the 2 cohorts have to be considered; for example, larger proportions of patients in need of bridging and not meeting ZUMA-1 eligibility, respectively, in the tisa-cel cohort. Moreover, although the PFS benefit of axi-cel remained significant after multivariable adjustment for confounders, there may be hidden biases in favor of axi-cel since the superior safety features and the management peculiarities of tisa-cel, in particular the option of disconnecting production from apheresis by local cryopreservation, may promote the enrichment of patients who are deemed more instable or frail in the tisa-cel group. This notion is supported by a survey among GLA SOC CAR users, suggesting that selection criteria favoring tisa-cel are primarily uncertainties regarding CAR T-cell indication and the outlook of CAR T-cell treatment if performed in the current patient state; but also higher age and comorbidities. In contrast, criteria favoring the selection of axi-cel comprise the necessity of a short vein-to-vein time and a large tumor mass, but also younger age and absence of comorbidity. The full survey can be found in the supplemental Appendix.
With 13 cases, the proportion of patients undergoing CAR T-cell therapy after a prior alloHCT was comparably large. Of note, NRM in this subset was significantly higher than in the alloHCT-naïve patients. However, this was based on only 3 events, and the CIs, therefore, were extremely wide. Most importantly, and in keeping with previous case series,40,41 PFS and OS of the alloHCT group was not inferior to that of the remainder, suggesting that a history of alloHCT should not preclude a subsequent CAR T-cell therapy in patients with LBCL if clinically indicated.
Being a retrospective registry report, our study has several limitations. There is certainly heterogeneity in patient selection across various centers. Data quality and granularity suffer from the retrospective nature of data collection. On the other hand, particular strengths of this analysis consist in the large sample size, enabling informative risk factor analyses, and the comprehensive coverage of the German SOC CAR T-cell therapy activity, with almost all qualified centers contributing data.
In conclusion, although this large registry analysis basically confirms the results of the pivotal trials, it highlights some particularities of CD19-directed CAR T-cell treatment of LBCL in the real-world setting. These include a relevant risk of delayed infection-related NRM raising implications for potential prophylaxis strategies, and the finding that effective bridging is an important predictor of CD19-directed CAR T-cell treatment efficacy and can overcome the adverse impact of actively proliferating disease on the outcome. Finally, our data suggest different safety/efficacy profiles of the 2 constructs available in the SOC setting, with less toxicity of tisa-cel, better disease control with axi-cel, and comparable survival. Before these differences can be considered for individualized product selection, however, they need to be substantiated in confirmative studies.
Authorship
Contribution: W.A.B. designed the research, collected and analyzed data, and wrote the manuscript; P.M. analyzed data and wrote and reviewed the manuscript; M. Schmitt, U.H., M. Subklewe, B.v.T., F.A., E.M.W.-D., G.G.W., R.M., O.P., U.S., C.K., M.v.B., M. Stelljes, B.G., C.D.B., V.V., D.M., M.T., M.A.F., R.S., L.B., P.B., V.B., J.H., C.H., S.T., D.W.B., C.L., and N.K. collected patient data and reviewed the manuscript; P.D. designed the research, collected and analyzed data, and wrote the manuscript.
Conflict-of-interest disclosure: W.A.B. received honoraria and travel funds from Gilead, Novartis, Miltenyi, and Janssen; and research grants from Miltenyi. M. Schmitt received funding for collaborative research from Apogenix, Hexal, and Novartis; travel grants from Hexal and Kite; financial support for educational activities and conferences from Bluebird Bio, Kite, and Novartis; is a board member for MSD and (co-)PI of clinical trials of MSD, GSK, Kite, and BMS, as well as co-founder and shareholder of TolerogenixX Ltd. U.H. received honoraria from Novartis, Gilead, BMS, Miltenyi, Roche. M. Subklewe received research support from Amgen, BMS, Gilead, Miltenyi, MorphoSys, Novartis, Roche, Seagen; sits on the advisory board for Amgen, BMS, Gilead, Janssen, Novartis, Pfizer, Seagen; received speaker’s bureau fees from Amgen, BMS, Gilead, Novartis, Pfizer, Takeda. B.v.T. is an advisor or consultant for BMS/Celgene, Incyte, Miltenyi, Novartis, Pentixafarm, Amgen, Pfizer, Takeda, Merck Sharp & Dohme, and Gilead Kite; received honoraria from AstraZeneca, Novartis, Roche Pharma AG, Takeda, and Merck Sharp & Dohme; received research funding from Novartis (Inst), Merck Sharp & Dohme (Inst), and Takeda (Inst); received travel support from AbbVie, AstraZeneca, Kite-Gilead, Merck Sharp & Dohme, Takeda, and Novartis. F.A. received honoraria from Novartis, Gilead, BMS, Janssen, Takeda, and Therakos/Mallinckrodt; received research funding from Therakos/Mallinckrodt. G.W. received honoraria Gilead, Novartis, Takeda, Clinigen, and Amgen. O.P. received honoraria or travel support from Astellas, Gilead, Jazz, MSD, Neovii Biotech, Novartis, Pfizer, and Therakos; received research support from Gilead, Incyte, Jazz, Neovii Biotech, and Takeda; and is on advisory boards of Jazz, Gilead, MSD, Omeros, Priothera, Shionogi, and SOBI. U.S. is on the advisory board for Novartis. C.K. is on advisory boards for Abbvie, Amgen, BMS, EusaPharm, GSK, Janssen, Kite/Gilead, Medigene, Novartis, Roche, Sanofi, Takeda, Pfizer, and Incyte. M.v.B. received honoraria and travel funds from Gilead, Novartis, Janssen, Takeda, and Daiichi Sankyō. M. Stelljes received honoraria from Kite/Gilead, Jazz, MSD, Novartis, Pfizer, and BMS/Celgene. C.D.B. received honoraria and travel funds from BMS, Amgen, Novartis, Jazz, Gilead, and Janssen. V.V. received honoraria and travel funds for Gilead; received honoraria and consultancies from Novartis, Gilead, and BMS/Celgene. D.M. received consultancies for AbbVie, Roche, BMS, Hexal, Novartis, MSD, Celgene, Janssen-Cilag, Pfizer, Astra-Zeneca, and Jazz Pharma. J.H. received honoraria from Merck Sharp & Dohme, AstraZeneca, Janssen, Amgen, Pfizer, Roche, Abbvie, Incyte, Jazz, and Bristol Myers Squibb. J.H. received honoraria from Merck Sharp & Dohme, AstraZeneca, Janssen, Amgen, Pfizer, Roche, Abbvie, Incyte, Jazz, and Bristol Myers Squibb. S.T. received honoraria and travel funds from Abbvie, BMS/Celgene, EUSA Pharma, Janssen, Gilead, Medigene, Novartis, and Pfizer; received research funding from BMS/Celgene and Gilead. N.K. received honoraria Kite/Gilead, Jazz, MSD, Neovii Biotech, Novartis, Riemser, Pfizer, and BMS; received research support from Neovii, Riemser, Novartis, and BMS. P.D. received consultancy from AbbVie, AstraZeneca, Bluebird Bio, Gilead, Janssen, Novartis, Riemser, and Roche; received speaker’s bureau fees from AbbVie, AstraZeneca, Gilead, Novartis, Riemser, and Roche; and received research support from Riemser. The remaining authors declare no competing financial interests.
The current affiliation for D.M. is Department of Hematology and Oncology, University Hospital Magdeburg, Magdeburg, Germany.
Correspondence: Wolfgang A. Bethge, University Hospital Tuebingen, Hematology and Oncology, Otfried-Mueller Str 10, D-72076 Tuebingen, Germany; e-mail: wolfgang.bethge@med.uni-tuebingen.de.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.