

TO THE EDITOR:
Bruton’s tyrosine kinase (BTK) is a common target for therapeutic intervention in patients with chronic lymphocytic leukemia (CLL).1,2 Ibrutinib is a first-generation BTK inhibitor (BTKi) that covalently binds to BTK to disrupt the B-cell receptor signaling pathway.3,4 Although BTKis are known to be an effective therapy for CLL, treatment-related toxicities can lead to the discontinuation of therapy.5,6 Acalabrutinib is a second-generation BTKi developed to reduce off-target toxicities.7,8
Though BTKis have improved the management of CLL, some patients experience clinical resistance and relapse during treatment due to mutations in arising clonal cell populations.9,10 This clonal evolution occurs when a dividing cell develops a new mutation that results in a greater competitive advantage compared with the surrounding cells.11-13 Clonal evolution can lead to treatment resistance when an expanding subclone contains a mutation that prevents the treatment agent from effectively targeting the malignant cells.14-16 Mutations that prevent BTKi binding, such as C481S BTK, or reconstitute B-cell receptor signaling independent of functional BTK, such as mutations in PLCG2, can drive treatment resistance in BTKi therapy.17-19 Identifying the evolution of subclones containing BTKi-resistant mechanisms before they expand provides an opportunity to alter the course of treatment before relapse occurs.
Although it has been shown that developing subclones with CLL-relevant mutations typically leads to negative outcomes during ibrutinib treatment, the follow-up time is often limited.9,14,20-22 Resistance mechanisms in acalabrutinib treatment have been investigated,23 but no study has been done to investigate the impact of clonal evolution on the response of patients treated with acalabrutinib. Here, we performed a longitudinal analysis of up to 8 years to better understand genomic heterogeneity and clonal evolution during ibrutinib and acalabrutinib treatments.
We collected blood samples from 38 CLL patients at multiple time points during their BTKi treatment (supplemental Table 1; supplemental Figure 1A). Prior to BTKi treatment, B cells were collected as a baseline measurement, and T cells were collected as a germline control. B-cell samples were then collected yearly for 3 years during treatment, with an additional sample taken at the most recent visit during the study period. We collected a median of 5 time points (range, 2-6) for each patient during their BTKi treatment (supplemental Figure 1B). For patients who relapsed, the final sample was collected at the time of relapse. Samples were sequenced using 200× whole-exome sequencing, providing deep sequencing data at each time point (supplemental Methods).
Our patient cohort includes 21 patients treated with ibrutinib and 17 treated with acalabrutinib. At the beginning of this study, 12 ibrutinib patients and 5 acalabrutinib patients were relapsed/refractory, whereas the rest were treatment naive (supplemental Results). Of the 38 patients, 19 continued to respond throughout the study, and 19 relapsed. Eight patients treated with ibrutinib developed Richter’s transformation. We found no significant difference in the progression-free survival for patients treated with acalabrutinib compared with those treated with ibrutinib (log-rank P = .11, supplemental Figure 2).
Understanding the clonal architecture of malignant B cells illuminates disease-causing or relapse-inducing mutations that may have undergone clonal expansion. Using variant allele frequencies (VAF) of the somatic mutations within each sample, we identified clonal evolution in 19 patients and no clonal evolution in the other 19 patients. Clonal evolution followed 3 general patterns: (1) new clone emergence (11/19 patients), where at least 1 new subclone emerged during treatment; (2) clonal selection (5/19 patients), where at least 1 subclone was selected against and decreased in prevalence while no new subclones emerged during treatment; and (3) clonal replacement (3/19 patients), where a new subclone appeared during treatment and replaced all subclones that descended from the founding clone (Figure 1A shows 1 example case per pattern). All 3 of these patterns of clonal evolution were seen in both BTKi cohorts.
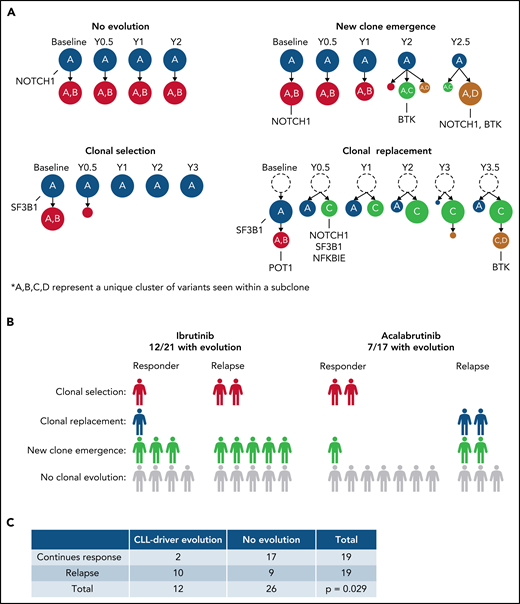
An overview of the clonal evolution identified in this study. (A) The patterns of clonal evolution identified, with an example of 1 patient from each pattern. This includes patient 28 (no evolution), patient 26 (new clone emergence), patient 27 (clonal selection), and patient 32 (clonal replacement). Gene names represent mutated CLL-relevant genes found within the indicated subclone. A, B, C, and D represent a unique cluster of variants seen within a given subclone that distinguishes it from other subclones. Richter’s transformation patients account for 4/19 of those with no evolution, 3/11 with new clone emergence, and 1/5 with clonal selection. (B) A breakdown of the clonal evolution found within each treatment cohort. Each cohort is separated into patients who continued to respond to the given BTKi and those who relapsed during BTKi treatment. (C) A Fisher’s exact test comparing the presence or absence of evolving subclones containing CLL-driver mutations to the treatment outcome of all patients.
An overview of the clonal evolution identified in this study. (A) The patterns of clonal evolution identified, with an example of 1 patient from each pattern. This includes patient 28 (no evolution), patient 26 (new clone emergence), patient 27 (clonal selection), and patient 32 (clonal replacement). Gene names represent mutated CLL-relevant genes found within the indicated subclone. A, B, C, and D represent a unique cluster of variants seen within a given subclone that distinguishes it from other subclones. Richter’s transformation patients account for 4/19 of those with no evolution, 3/11 with new clone emergence, and 1/5 with clonal selection. (B) A breakdown of the clonal evolution found within each treatment cohort. Each cohort is separated into patients who continued to respond to the given BTKi and those who relapsed during BTKi treatment. (C) A Fisher’s exact test comparing the presence or absence of evolving subclones containing CLL-driver mutations to the treatment outcome of all patients.
Of the 21 patients who received ibrutinib, 12 (57.1%) had clonal evolution during treatment. This includes 8 patients with a pattern of new clone emergence, 3 with clonal selection, and 1 with clonal replacement during treatment (Figure 1B). Clonal evolution was observed in 7 (41.2%) of the 17 patients who received acalabrutinib treatment, including 3 with new clone emergence, 2 with clonal selection, and 2 with clonal replacement. These findings show that patients treated with acalabrutinib develop similar patterns of clonal evolution as those treated with ibrutinib.
We investigated the impact of evolving subclones containing CLL-relevant mutations on treatment outcomes (supplemental Table 2). Of the 19 patients who relapsed, 9 (47.4%) had evolving mutations (change in VAF ≥0.1) in CLL-relevant genes. Only 2 of the 19 patients (10.5%) who responded to treatment throughout this study had evolving mutations in CLL-relevant genes. This data shows a significant association between the evolution of subclones containing mutations in CLL-relevant genes and poor treatment outcomes (Fisher P = .029, Figure 1C). As TP53 mutations are known to be associated with poor clinical outcomes regardless of treatment and are highly associated with Richter’s transformation,24,25 we separated patients with baseline TP53 mutations to account for potential bias (supplemental Figure 3). When only including patients without confounding baseline TP53 mutations (27 patients; 8 relapsed, 19 responded), we found that 5 of the remaining 8 patients who relapsed had evolving subclones with CLL-relevant mutations, compared with only 1 of the 19 patients who responded. This analysis reveals a strong association between evolving CLL-relevant mutations and relapse (Fisher P = .011).
We then determined whether the evolution of CLL-relevant mutations can be used as a prognostic marker. Setting a landmark at year 2 of treatment, we grouped patients based on the presence or absence of at least 1 evolving subclone with a CLL-relevant mutation up to that point. Comparing patients with this evolution in the first 2 years to those without, we saw a significant difference in the progression-free survival beyond year 2 between the 2 groups (log-rank P = .0004, Figure 2). This same significant difference was observed when looking solely at the ibrutinib cohort (log-rank P = .0021, supplemental Figure 4A) or the acalabrutinib cohort (log-rank P = .04, supplemental Figure 4B). We concluded that patients with evolving CLL-relevant mutations in the first 2 years of treatment are more likely to relapse while receiving BTKi treatment.
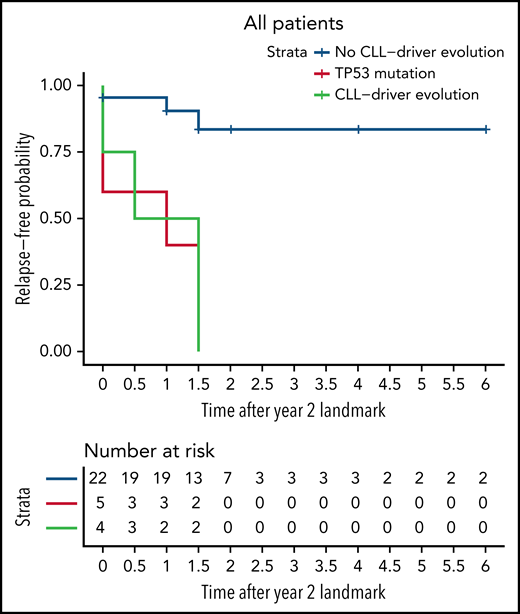
Kaplan-Meier curve showing the probability of relapse-free survival over time. A landmark was set at year 2 to group patients with or without the evolution of subclones containing CLL-relevant mutations up to that point. The red line includes any patient with baseline TP53 mutations, the green line includes patients without baseline TP53 mutations who had at least one evolving CLL-relevant mutation in the first 2 years of treatment, and the blue line includes patients without baseline TP53 mutations or evolving CLL-relevant mutations at year 2. The x-axis represents the relapse-free survival time since the second year of treatment. There is a significant difference in the relapse-free survival of patients who do not have CLL-relevant evolution in the first 2 years compared with those who do (log-rank P = .0004 when comparing the green and blue lines).
Kaplan-Meier curve showing the probability of relapse-free survival over time. A landmark was set at year 2 to group patients with or without the evolution of subclones containing CLL-relevant mutations up to that point. The red line includes any patient with baseline TP53 mutations, the green line includes patients without baseline TP53 mutations who had at least one evolving CLL-relevant mutation in the first 2 years of treatment, and the blue line includes patients without baseline TP53 mutations or evolving CLL-relevant mutations at year 2. The x-axis represents the relapse-free survival time since the second year of treatment. There is a significant difference in the relapse-free survival of patients who do not have CLL-relevant evolution in the first 2 years compared with those who do (log-rank P = .0004 when comparing the green and blue lines).
Within our cohort, 3 patients treated with ibrutinib and 3 treated with acalabrutinib developed a subclone containing a C481S BTK mutation, with VAFs ranging from 0.05 to 0.8. Each of these patients relapsed in no more than 3.5 years. The C481S mutation was detectable 1 year before relapse in 2 patients and only detectable at relapse in the remaining 4 patients. Additional information about the BTK mutations in each patient can be found in supplemental Results. Like ibrutinib, we found that acalabrutinib is capable of driving the evolution of subclones containing BTK mutations. Notably, we did not detect any PLCG2 mutations within our cohort.
In conclusion, we have determined that acalabrutinib treatment leads to the development of similar clonal evolution patterns as observed in ibrutinib treatment (supplemental Discussion). Additionally, the evolution of subclones with CLL-relevant mutations in the first 2 years of treatment is significantly associated with eventual relapse during either BTKi treatment. We also find that baseline TP53 somatic mutations or the development of subclones containing BTK mutations increases relapse risk in both BTKi treatments. Within a clinical context, our findings can provide a subclone-level understanding of a patient’s response to ibrutinib or acalabrutinib and help identify markers for potential relapse.
Acknowledgments
The support and resources from the Center for High-Performance Computing at the University of Utah are gratefully acknowledged. We are grateful to the patients who provided tissue samples for these studies to the OSU Comprehensive Cancer Center Leukemia Tissue Bank Shared Resource (supported by NCI P30 CA016058). We also thank the OSU Leukemia Tissue Bank for assistance with the CLL samples.
G.S.B., X.H., Y.Q., and G.T.M. were supported by National Institute of Health (NIH), National Cancer Institute (NCI) grant U24 CA209999 and NIH, National Human Genome Research Institute (NHGRI) grant R01 HG009000 both awarded to G.T.M. D.M.S. was awarded and supported by NIH, NCI award K23 CA212271. J.A.W and K.A.R. are Scholars in Clinical Research of The Leukemia and Lymphoma Society. J.A.W. was awarded and supported by NIH, NCI grants R01 CA177292 and R01 CA197870. Research reported in this publication used the High-Throughput Genomics and Bioinformatic Analysis Shared Resource at Huntsman Cancer Institute at the University of Utah and was supported by the NIH, NCI award P30 CA042014. The NIH Shared Instrumentation Grant 1S10OD021644-01A1 partially funded the computational resources used for this study.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Authorship
Contribution: G.S.B. contributed to the genomic data processing and analysis, variant identification, subclone analysis, and results interpretation; X.H. contributed to the study design, project coordination, sample processing for DNA sequencing, data analysis, and interpretation; Y.Q. contributed to the data processing, subclone analysis, and interpretation; S.M. and S.T. contributed to the sample processing for DNA sequencing; K.A.R., J.A.W., and J.C.B. contributed to clinical oversight and patient sample selection; D.M.S., J.A.W., J.C.B., G.T.M., and X.H. conceived the project; D.M.S. and J.A.W. contributed to the project coordination, clinical oversight, and sample collection and transfer; G.S.B., X.H., Y.Q., G.T.M., D.M.S., and J.A.W. contributed to manuscript writing; and all authors contributed to the manuscript refinement and read and approved the final manuscript.
Conflict-of-interest disclosure: J.A.W has received research funding from Pharmacyclics, Janssen, Karyopharm, Morphosys, and Schrodinger and consults for Janssen, Pharmacyclics, Abbvie, AstraZeneca, Beigene, Loxo, and Newave. D.M.S. has received research funding from Acerta Pharma, Gilead Sciences, Karyopharm Therapeutics, Mingsight, Arqule, Novartis, Verastem, and Juno Therapeutics, and she has received consulting fees from Pharmacyclics/Janssen, Karyopharm Therapeutics, Beigene, Innate, AstraZeneca, Abbvie, CSL Behring, Celegene, TG Therapeutics, and Innate Pharma. K.A.R. receives research funding from AbbVie, Genentech, and Janssen; consulted for Pharmacyclics, AstraZeneca, Actera Pharma, Genentech, AbbVie, Beigene, and Innate Pharma; and received travel funding from AstraZeneca. The remaining authors declare no competing financial interests.
Correspondence: Gabor T. Marth, 15 North 2030 East, Room 7410B, Salt Lake City, UT 841121; e-mail: gabor.marth@gmail.com; Deborah M. Stephens, 2000 Circle of Hope, Research South 5509, Salt Lake City, UT 84112; e-mail: deborah.stephens@hci.utah.edu; and Jennifer A, Woyach, 455D Wiseman Hall, 410 West 12th Avenue, Columbus, OH 43210; e-mail: jennifer.woyach@osumc.edu.
VCF files containing the somatic mutation information for each patient, the custom scripts used, and the processed datasets generated and analyzed in the study are available in the GitHub repository at https://github.com/gageblack/CLL-clonal-evolution.
Send data sharing requests via e-mail to the corresponding author.
The online version of this article contains a data supplement.

