

In this issue of Blood, Kapeni et al1 reported that the deletion of p57Kip2 leads to increased proliferation of neural crest cell–derived sympathoadrenal (SA) progenitors in the mouse embryonic aorta-gonad-mesonephros (AGM) region, which in turn generates more SA cells that promote the repopulation capacity of hematopoietic stem cells (HSCs) by secreting catecholamines.
A method that facilitates the de novo generation of human HSCs from pluripotent stem cells (PSCs) would undoubtedly have a profound impact on the fields of hematopoietic biology and regenerative medicine. However, the ability to produce bona fide HSCs in a dish remains elusive. Thus, it is necessary to gain a better understanding of embryonic hematopoiesis. In recent years, single-cell RNA sequencing (scRNA-seq) technology has substantially improved our knowledge of hematopoietic ontogeny in both humans and mice.2-4 But much less is known about the niche components and signaling modules in the AGM region where pre-HSCs emerge in a process referred to as the endothelial-to-hematopoiesis transition.
In the study by Kapeni et al, the authors discovered that deletion of the cell cycle regulator p57Kip2 increases the repopulation capacity of mouse HSCs at embryonic day 12 (E12) in the AGM region and at E11 in fetal liver but fails to do so at E11 and E12 in both the placenta and yolk sac, indicating that p57Kip2 has a tissue-specific effect on HSC development. p57Kip2 has been reported to be indispensable for the quiescence and maintenance of adult HSCs.5,6 In contrast to high expression in adult HSCs, AGM hematopoietic stem and progenitor cells exhibited little to no expression of p57Kip2, which may account for their active cell cycling status. Of note, p57Kip2-deficient AGM HSCs exhibited robust repopulation capacity even 4 months after being transplanted into adult mice. Conversely, p57Kip2-deficient adult HSCs exhibited an exhausted phenotype. Mascarenhas et al7 previously found that AGM HSCs retain their immature characteristics to some extent after transplantation into adult mice, a finding that opposes the idea that the adult bone marrow niche can facilitate maturation of nascent HSCs. This finding suggests a scenario regarding the induction of PSCs to HSCs in the sense that once nascent HSCs are successfully generated from PSCs, additional culture mimicking the fetal liver microenvironment will be indispensable for nascent HSCs to mature to adult HSCs. Elucidating the molecular mechanisms that allow AGM HSCs to maintain proliferation without becoming exhausted after transplantation into adult mice would likely have a significant impact on the ex vivo expansion of adult HSCs.
However, p57Kip2 was mainly detected in Ngfr+ SA cells and existed at much lower levels in CD34+ endothelial cells and Pdgfrß+ mesenchymal cells. This implies that p57Kip2 probably affects AGM HSCs in a way that is not cell autonomous, although a conditional knockout mouse model is needed to completely rule out the direct effect of p57Kip2 on HSCs. As a result, deletion of p57Kip2 significantly increased the population of Ngfr+ cells in the AGM region and subsequently generated higher levels of catecholamines, which positively regulate AGM HSCs by binding to the β2-adrenergic receptor. Furthermore, the expression of adrb2 (encoding β2-adrenergic receptor) was high in pre-HSC II, but low in pro-HSCs and pre-HSC I. This suggests that pre-HSC II should be the main population responsible for catecholamines. Thus, the question becomes, what is the downstream signaling pathway of β2-adrenergic receptor and the resulting biological effect on pre-HSC II (eg, does it increase proliferation or promote maturation of pre-HSC II)? It would be interesting to investigate whether catecholamines can also expand or maturate human nascent HSCs induced from PSCs, if any.
Next, the authors performed scRNA-seq on Ngfr+ cells isolated from the AGM region and defined a classic SA differentiation pathway by which SA cells arise from neural crest cells via SA progenitors. Among these SA lineage cells, SA progenitors are the major population expressing p57Kip2, suggesting that deletion of p57Kip2 had the most prominent effect on SA progenitors. In addition, Kapeni et al found that downregulation of Notch signaling is required for neural crest cells to commit to SA cells. Of note, attenuation of Notch signaling is also necessary for the endothelial-to-hematopoietic transition, which occurs at a similar time window (E10.5). It would be interesting to further investigate the cellular source of this Notch signaling inhibition in the AGM region. Given that Notch signaling requires cell-cell interaction, the spatial location of these cells may play a critical role in this scenario.
Interestingly, analysis of cell lineage inference also revealed a second differentiation pathway by which mesenchymal cells arise from neural crest cells. Of note, these mesenchymal cells are highly enriched in the ventral part of the aorta where endothelial-to-hematopoietic transition occurs. Whether and at which stage these mesenchymal cells play a role in HSC development needs to be examined. Interestingly, analysis of preliminary scRNA-seq data showed several potential ligand-receptor interactions between mesenchymal cells and AGM HSCs. The cellular origin of this mesenchymal cell cluster requires further investigation because the somatic mesoderm marker Mexo1 was also expressed in a subset of this cluster.
Overall, the authors revealed that p57Kip2 deficiency expanded SA progenitors in the AGM region and subsequently increased SA cells, which positively regulate AGM HSCs via the catecholamine–β2-adrenergic receptor axis. A cluster of neural crest cell–derived mesenchymal cells enriched in the ventral part of aorta was also discovered. The influence of these mesenchymal cells on the development of AGM HSCs awaits further investigation (see figure).
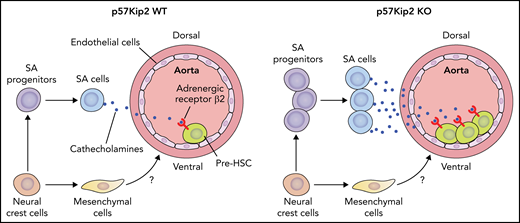
Deletion of p57Kip2 increases the proliferation of neural crest cell–derived SA progenitors in the E11 mouse AGM region. The expanded SA progenitors in turn produce more SA cells, which promote the repopulation capacity of hematopoietic stem cells by the catecholamine–β2-adrenergic receptor signaling axis. The neural crest cells also give rise to mesenchymal cells enriched in the ventral part of the aorta in the AGM region. The role of these mesenchymal cells in embryonic hematopoiesis awaits investigation. Professional illustration by Patrick Lane, ScEYEnce Studios.
Deletion of p57Kip2 increases the proliferation of neural crest cell–derived SA progenitors in the E11 mouse AGM region. The expanded SA progenitors in turn produce more SA cells, which promote the repopulation capacity of hematopoietic stem cells by the catecholamine–β2-adrenergic receptor signaling axis. The neural crest cells also give rise to mesenchymal cells enriched in the ventral part of the aorta in the AGM region. The role of these mesenchymal cells in embryonic hematopoiesis awaits investigation. Professional illustration by Patrick Lane, ScEYEnce Studios.
Although several cell types have been identified as the potential niche for AGM HSCs,8-10 the complexity of both the cellular components and signaling modules in the AGM region is far from fully understood. Given that the stagewise specification, emergence, and maturation of HSCs happens exclusively in the ventral part of the aorta in the AGM region, analyzing single-cell-resolved spatial transcriptomics for specific embryonic stages in the AGM region should be able to uncover the polarized niche components and signaling modules essential for embryonic hematopoiesis. In addition, multiomics sequencing of AGM hemogenic endothelial cells and HSCs would provide deeper insights into hematopoietic ontogeny from the perspective of transcriptomics, chromatin accessibility, and proteomics. A thorough understanding of the intrinsic hematopoietic program and extrinsic niche cells and the signals involved in the regulation of AGM HSCs will pave the way for the in vitro generation of HSCs from PSCs, which is still a significant barrier to be overcome in the field of hematopoiesis.
Conflict-of-interest disclosure: The authors declare no competing financial interests.

