

In this issue of Blood, Carratt et al1 show that mutant SETBP1 induces a MYC-driven transcriptional program that promotes leukemogenesis in CSF3R mutant leukemia. Execution of this program is dependent on lysine-specific demethylase 1 (LSD1), suggesting that combining inhibitors of JAK2 and LSD1 may be effective in treating chronic neutrophilic leukemia (CNL).
CNL and atypical chronic myeloid leukemia (aCML) are aggressive myeloproliferative neoplasms (MPNs) of older individuals with a median survival of 2 to 3 years and no curative drug therapy. Both are characterized by granulocytosis, including a sizable proportion of neutrophils, and lack genetic abnormalities that are diagnostic of other MPN entities, such as BCR-ABL1 of CML. The discovery of activating mutations in CSF3R, also known as granulocyte-colony stimulating factor (G-CSF) receptor, in most CNL and a variable fraction of aCML cases has provided an explanation for their neutrophilic differentiation bias and established a molecular diagnostic marker.2 Future disease classifications may list CSF3R mutant MPN as a distinct entity, much in the way that the presence of BCR-ABL1 establishes a diagnosis of CML, irrespective of the specific MPN morphology.
The most common CSF3R mutation, CSF3RT618I, is localized in the membrane-proximal portion of the extracellular domain, leads to constitutive JAK/STAT signaling, and induces a CNL-like leukemia in mice.2 A subset of patients with CNL has a second CSF3R mutation that truncates the distal part of the intracellular domain and promotes activation of SRC family kinases.2 Compared with CML, CNL is genetically more complex. At diagnosis, almost all patients exhibit additional mutations in genes associated with myeloid malignancies, including ASXL1, TET2, EZH2, and SETBP1. Although SETBP1 mutations occur across the spectrum of myeloid neoplasms, they have a particularly strong association with aCML and CNL.3 SETBP1 was reported to promote oncogenesis either by inhibiting PP2A, a widely expressed serine/threonine phosphatase with tumor suppressor function,4 or by recruiting transcriptional repressors to critical myeloid differentiation genes such as RUNX1.5 Oncogenic SETBP1 mutations localize to the β-TrCP1 degron motif and impair binding to SCF1 E3 ligase complexes, preventing proteasomal SETBP1 degradation. SETBP1 mutations in myeloid malignancies overlap with germline mutations in patients with Schinzel-Giedion syndrome, a developmental disorder associated with physical stigmata and intellectual disability. In several myeloid malignancies, including acute myeloid leukemia (AML), SETBP1 mutations or high SETBP1 expression are associated with poor survival.
Ruxolitinib, a JAK2 inhibitor, has shown modest activity in patients with CSF3R mutant CNL and aCML. Although data are sparse, patients with less complex genetics seem to achieve better responses, suggesting that targeting pathways activated by secondary mutations is required to improve outcomes.6 Considering the strong association between CSF3R and SETBP1 mutations, Carrat et al. studied the molecular and cellular consequences of combining CSF3RT618I and SETBP1D868N, the most common SETBP1 mutation, using in vitro and in vivo models. Unlike CSF3RT618I alone, CSF3RT618I plus SETBP1D868N transformed hematopoietic cells to growth factor independence, conferred serial colony replating capacity (an indicator of self-renewal), and induced a rapidly fatal, neutrophil-predominant MPN in mice. Coexpression of SETBP1D868N impaired myeloid maturation of progenitor cells either expressing CSF3RT618I or cultured with G-CSF, suggesting that SETBP1D868N inhibits myeloid-lineage differentiation induced by G-CSF signaling. Interestingly, overexpression of wild-type SETBP1 partially recapitulated the SETBP1D868N phenotype, suggesting a gene dosage effect. RNA sequencing and chromatin occupation studies revealed activation of a strong MYC-driven transcriptional program with upregulation of genes normally expressed in hematopoietic stem cells, including Hoxa9, Hoxa10, and Meis1. Extensive overlap between H3K4me3 and H3K27Ac peaks and MYC targets implicated MYC as the master regulator of the SETBP1D868N signature in CSF3RT618I mutant cells. An inhibitor screen in myeloid progenitors immortalized by coexpression of CSF3RT618I and SETBP1D868N revealed profound dependence on LSD1. LSD1 inhibitors, particularly ORY-1001, reduced MYC expression and partially reversed the MYC-driven aberrant transcriptional program. When combined with ruxolitinib, ORY-1001 prologed survival of mice with SETBP1D868N/CSF3RT618I-induced leukemia, suggesting therapeutic utility (see figure).
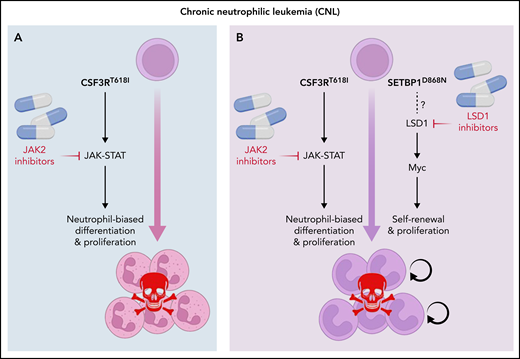
Combining JAK2 and LSD1 inhibitors in SETBP1 mutant CNL. (A) JAK2 inhibitors are relatively effective only in the rare CNL cases with isolated CSF3R mutations. (B) Combining JAK2 inhibitors with LSD1 inhibitors may be effective in SEPBP1 mutant CNL.
Combining JAK2 and LSD1 inhibitors in SETBP1 mutant CNL. (A) JAK2 inhibitors are relatively effective only in the rare CNL cases with isolated CSF3R mutations. (B) Combining JAK2 inhibitors with LSD1 inhibitors may be effective in SEPBP1 mutant CNL.
What’s next? The authors previously reported that SETBP1D868N enhances MAPK signaling in cells transformed by NRASK12D, but this does not seem to be associated with increased MYC activity, suggesting that SETBP1 recruits transcriptional cofactors in a cell context-dependent manner.7 In fact, recent data show that recruitment of KMT2A (previously mixed-lineage leukemia) is required for SETBP1-mediated myeloid transformation, consistent with SETBP1 functioning as a transcriptional hub.8,9 Determining the composition of SETBP1 containing transcription complexes using an unbiased proteomics-based approach may reveal additional therapeutic opportunities. Based on the work of Carrat et al, one may suspect that such cofactors will differ based on the upstream signaling input. Although they clearly demonstrate that LSD1 is critical for CSF3RT618I/SETBP1D868N–driven leukemia, it remains to be determined how exactly SETBP1 and LSD1 interact. However, irrespective of the precise mechanism, LSD1 inhibitors and ruxolitinib are strongly synergistic in vitro. Given the dismal prognosis of patients with CNL and aCML, a clinical trial combining ruxolitinib with an LSD1 inhibitor is clearly warranted. One may speculate that the long-term results of this combination may hinge on whether and how fast the leukemia cells are able to rewire. A recent report showed that AML cells challenged with an LSD1 inhibitor activate MAPK signaling,10 suggesting that patients with CSF3RT618I mutant leukemia treated with a combination of JAK2 and LSD1 inhibitors may develop resistance by activating the RAS pathway and acquire sensitivity to mitogen-activated protein kinase kinase inhibitors.
The work of Carratt et al emphasizes that somatic mutations in cancer co-occur for a reason, providing a competitive solution to a molecular problem that manifests as a highly aggressive malignancy. On the bright side, this can lead to unexpected vulnerabilities that lend themselves to rational therapeutic exploitation. Perhaps there is the light at the end of the tunnel for patients with CNL.
Conflict-of-interest disclosure: The authors declare no competing financial interests.

