TO THE EDITOR:
Evans syndrome (ES) is a rare condition, defined as the presence of 2 autoimmune cytopenias: more frequently, autoimmune hemolytic anemia (AIHA) and immune thrombocytopenia (ITP) and, rarely, autoimmune neutropenia (AIN).1-3 ES can be classified as primary or secondary to various conditions, including lymphoproliferative disease, primary immune deficiencies, or other systemic autoimmune diseases. The onset of ES may be acute and life threatening, whereas its clinical course is usually chronic and marked by several relapses of AIHA, ITP, or both. In addition, risks of thrombosis and infections were ∼2 times those observed in primary AIHA or ITP in a recent European study.3 First-line therapy is based on steroids, with or without intravenous immunoglobulins (IVIGs), followed by a second-line treatment largely depending on the cytopenia, and include rituximab, splenectomy, and cytotoxic immunosuppressants. Since their arrival, thrombopoietin receptors agonists (TPO-RAs), such as romiplostim and eltrombopag, have been increasingly used in primary ITP with high efficacy, and a good safety profile.4 However, their use in ES has never been systematically studied, and only a few case reports are available for this challenging population.5-12 In this observational study, we evaluated the efficacy and safety of TPO-RAs in a multicentric cohort of patients with ES.
We retrospectively evaluated patients with diagnosed ES and observed from June 1987 at 10 European tertiary hematologic hospitals (5 in Italy, 2 in Denmark, 1 in the United Kingdom, and 2 in Spain), who had received at least 1 TPO-RA agent for ITP. ES was diagnosed based on the concomitant or sequential onset of at least 2 immune cytopenias (ITP, AIHA, and/or AIN), each diagnosed according to current guidelines.4,13,14 The study was conducted according to the Declaration of Helsinki and approved by the local ethics committee. Response rates were evaluated at 1, 3, 6, and 12 months, and classified as partial (PR) or complete (CR) response, for platelets (PLTs) >30 × 109/L or >100 × 109/L, respectively. Emergent treatment adverse events (TAEs) were registered and graded according to Common Terminology Criteria for Adverse Events. In a second analysis, we compared baseline features, response to TPO-RAs, TAEs, and outcome in an ES cohort with a control group of 87 patients with primary ITP treated with TPO-RAs at the center of the first and senior authors BF and WB.
As shown in Table 1, 29 patients with ES (19 men and 10 women; median age, 61 years; range, 24-88) were included; 8 had ES secondary (28%) to lymphoproliferative disease (n = 2, 1 subjected to hematopoietic stem cell transplant), primary immune deficiencies (n = 2), antiphospholipid syndrome (APS; n = 3), and autoimmune diseases (n = 1). Regarding cytopenia type, all patients had ITP plus AIHA (n = 25) or AIN (n = 2) or all 3 (n = 2). The first presenting cytopenia was ITP in the majority of cases (62%), with positive anti-PLT autoantibodies in 45% of tested patients. All AIHA had a positive direct antiglobulin test for IgG (n = 17), IgG+complement at low titer (n = 9), and complement alone (n = 1). Bone marrow evaluation had been performed before TPO-RA treatment in 22 cases (85%) and mainly showed normocellularity or hypercellularity (77%) and increased megakaryocytes (86%), with dysmegakaryopoiesis and reticulin fibrosis (grade MF-0) in one-third of cases. A lymphoid infiltrate was noted in 14 cases, mainly polyclonal T cells or mixed B cells and T cells (93%), exceeding 10% only in 2 patients with B-cell lymphoma.
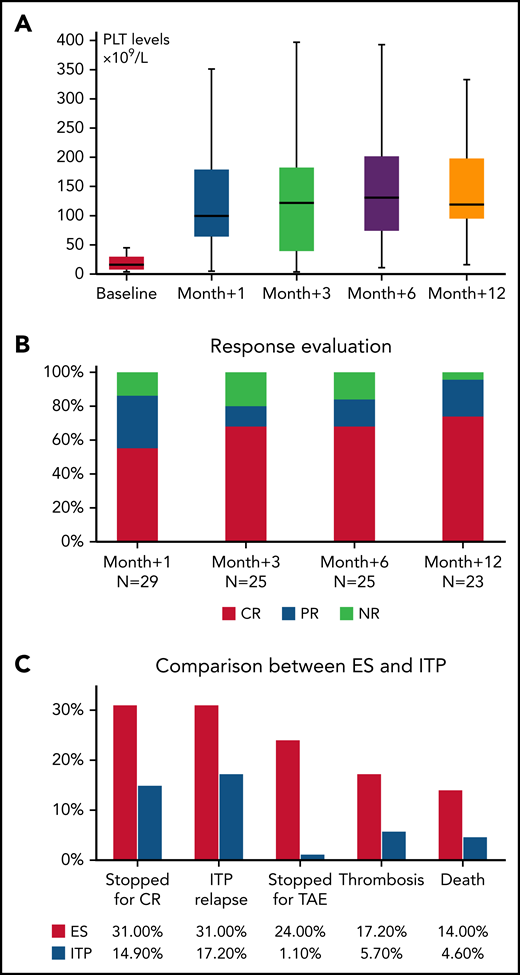
All patients had received treatment for ITP (median, 6 lines; range, 1-8), including steroids with or without IVIG in almost all cases (96%), rituximab (59% of patients), cytotoxic immunosuppressors (cyclosporine, mycophenolate, and azathioprine; 34%), splenectomy (26%), and danazol (11%). Responses are shown in Table 1. The median time from diagnosis to beginning treatment with a TPO-RA was 20 months (range, 1-1381), and treatment indication was nonresponse to previous or ongoing therapy in all cases. Twenty-three patients received eltrombopag (79%) and 6 romiplostim (21%), with a median maximal dose of 75 mg/d (50-150) and 3 μg/kg per week (range, 2-10), respectively. PLT values showed a progressive increase (Figure 1A), resulting in response rates (CR+PR) of 86% at month +1, 80% at month +3, 84% at month +6 and 96% at month +12 (Figure 1B). Of note, 86% of patients were receiving concomitant therapies at the time of initial treatment with a TPO-RA, including steroids with or without IVIG (n = 19), rituximab (n = 5), immunosuppressants (n = 3), and danazol (n = 4). Overall, the median duration of therapy was 12 months (1-71), and 10 patients were able to stop TPO-RA treatment for persistent CR (ie, treatment-free remission); however, 8 subsequently relapsed and required TPO-RA rechallenge (median relapse-free survival, 6 months; range, 3-66). Additional reasons for cessation of TPO-RA were nonresponse (n = 4) and TAEs (thrombosis, n = 3; thrombocytosis, n = 3; and increased bone marrow fibrosis MF-2, n = 1). Interestingly, 5 subjects were switched to an alternative TPO-RA (3 romiplostim to eltrombopag and 2 vice versa): 2 because of nonresponse and 3 for relapse. Of those, 3 patients responded. In addition, during TPO-RA treatment 10 subjects required additional rescue therapies because of intercurrent decrease in PLTs (steroids with or without IVIG, n = 4; cytotoxic immunosuppressants, n = 3; danazol, n = 1; dapsone, n = 1; and splenectomy, n = 1).

Evaluation of TPO-RA treatment in patients with ES and in a cohort of patients with ITP. (A) PLT counts during treatment in ES. (B) Response rates at various time points in ES. (C) Comparison of outcomes of ES with those of primary ITP. Response rates in ITP were 77% at month +1, 80% at month +3, 88% at month +6, and 92% at month +12.
Evaluation of TPO-RA treatment in patients with ES and in a cohort of patients with ITP. (A) PLT counts during treatment in ES. (B) Response rates at various time points in ES. (C) Comparison of outcomes of ES with those of primary ITP. Response rates in ITP were 77% at month +1, 80% at month +3, 88% at month +6, and 92% at month +12.
Eleven patients (38%) developed at least 1 TAE: grade 1 (G1) PLT fluctuations (n = 3), G2 bone marrow fibrosis (n = 2), G3 and G4 thrombosis (6 events in 5 patients: 2 pulmonary embolisms, 1 splanchnic thrombosis, 1 atrial thrombus, 1 cerebral vein thrombosis, and 1 myocardial infarction in a patient with APS), and G5 infection not related to a TPO-RA (n = 1). Of note, patients who underwent splenectomy had a higher, but not significant, frequency of thrombosis vs those who did not (28% vs 14%). In addition, 1 pulmonary embolism occurred during active hemolysis and concomitant treatment with prednisone 1 mg/kg per day. Finally, 4 patients died of infectious events (all men with multitreated primary ES: 56, 57, 78, and 87 years of age).
By comparing treatment outcomes of the 29 patients with ES with those in a cohort of 87 TPO-RA–treated patients with primary ITP, similar response rates were observed at all time points. Of interest, patients with ES had a higher treatment-free remission rate (P < .001) enabling them to discontinue TPO-RAs, but there was also a trend toward greater relapse of thrombocytopenia (P = .07). In addition, patients with ES showed a higher frequency of G3 and G4 TAEs (P < .001), particularly thrombotic events (P = .04), and they more often discontinued treatment because of TAEs (P < .001; Figure 1C).
In this study, we evaluated for the first time the efficacy and safety of TPO-RAs in patients with ES and observed response rates exceeding 80%, even in heavily pretreated patients. A switch from 1 TPO-RA to an alternative agent was also feasible, similar to that already reported for primary ITP.15 However, the great majority of patients with ES (>70%) required combination therapy, at variance with patients with primary ITP (<10% of cases). Moreover, approximately one-third of patients with ES experienced a PLT decrease during TPO-RA or an ITP relapse after discontinuation. These observations suggest a deeper immune dysregulation in ES that is not completely compensated, even by bone marrow stimulation with TPO-RA. Recently, we have consistently reported the more severe clinical course of ES as compared with primary cytopenias, with a remarkable number of relapses, multitreatments, and dismal outcomes.3 Importantly, TPO-RA use in ES was complicated by TAEs in approximately one-third of cases, particularly thrombosis in nearly 20% of subjects (approximately threefold that of primary ITP). In fact, patients with ITP are considered at higher risk of thromboembolism related to the high fraction of “young PLTs,” the augmented PLT and red cell microparticles, an increased resistance to protein C, and the augmented levels of plasminogen activator inhibitor-1,16 and the hazard may increase in patients receiving TPO-RA treatment.17 Additional risk factors may be simultaneous AIHA (present in 1 patient), where thrombotic risk has been related to intravascular hemolysis, anemia severity, and multiple treatments, particularly splenectomy.18-20 One patient with ES consistently developed pulmonary embolism during active hemolysis treated with high-dose steroids. Finally, underlying predisposing thrombophilic conditions, such as APS, should be assessed.3 Again, the 2 patients with ES with APS displayed severe thrombotic events.
We acknowledge that our study is limited by the small number of patients and the retrospective design; in addition, patients with ES were treated from 2013 through 2021, and the use of TPO-RA was optimized in the last 10 years regarding dosing, PLT target, and possibility of tapering and discontinuing the drug. However, we observed a homogeneous distribution of responses and TAEs, particularly thrombosis, throughout the follow-up, suggesting that our data provide a reliable snapshot of TPO-RA use in ES.
In summary, the data suggest that TPO-RAs in adult patients with ES are highly effective. However, careful monitoring is necessary to avoid thrombocytosis/PLT fluctuations and thromboses and to tune concomitant/rescues therapies. Finally, administration of TPO-RAs in patients with ES with an associated thrombophilic condition (ie, antiphospholipid syndrome and paroxysmal nocturnal hemoglobinuria) may be reconsidered, particularly with the advent of novel drugs (eg, spleen tyrosine kinase inhibitors and antineonatal Fc receptors) for ITP. Overall, ES confirms a more severe clinical pattern and profound immune dysregulation and thromboinflammation, as compared with primary ITP.
Acknowledgment
Funding support for this article was provided by the Italian Ministry of Health Current Research Grant.
Authorship
Contribution: B.F., N.C., and W.B. conceived the study and wrote the article; and all authors followed up on the patients, collected the data, and revised the article for important intellectual content.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Bruno Fattizzo, Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Via F. Sforza 35, 20122 Milan, Italy; e-mail bruno.fattizzo@unimi.it.
Original data are available upon e-mail request to the corresponding author (bruno.fattizzo@unimi.it).