Key Points
Sutimlimab reduced hemolysis, anemia, and fatigue in patients with CAD without transfusion requirement.
Sutimlimab treatment was generally well tolerated, with adverse events consistent with an older and medically complex patient population.

Abstract
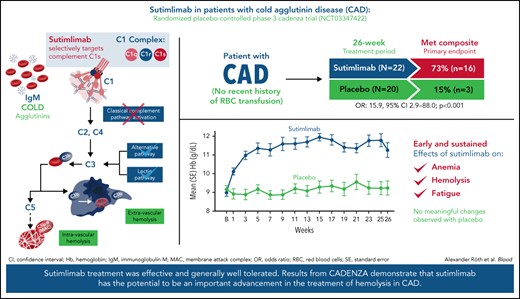
Sutimlimab, a first-in-class humanized immunoglobulin G4 (IgG4) monoclonal antibody that selectively inhibits the classical complement pathway at C1s, rapidly halted hemolysis in the single-arm CARDINAL study in recently transfused patients with cold agglutinin disease (CAD). CADENZA was a 26-week randomized, placebo-controlled phase 3 study to assess safety and efficacy of sutimlimab in patients with CAD without recent (within 6 months prior to enrollment) transfusion history. Forty-two patients with screening hemoglobin ≤10 g/dL, elevated bilirubin, and ≥1 CAD symptom received sutimlimab (n = 22) or placebo (n = 20) on days 0 and 7 and then biweekly. Composite primary endpoint criteria (hemoglobin increase ≥1.5 g/dL at treatment assessment timepoint [mean of weeks 23, 25, 26], avoidance of transfusion, and study-prohibited CAD therapy [weeks 5-26]) were met by 16 patients (73%) on sutimlimab, and 3 patients (15%) on placebo (odds ratio, 15.9 [95% confidence interval, 2.9, 88.0; P < .001]). Sutimlimab, but not placebo, significantly increased mean hemoglobin and FACIT-Fatigue scores at treatment assessment timepoint. Sutimlimab normalized mean bilirubin by week 1. Improvements correlated with near-complete inhibition of the classical complement pathway (2.3% mean activity at week 1) and C4 normalization. Twenty-one (96%) sutimlimab patients and 20 (100%) placebo patients experienced ≥1 treatment-emergent adverse event. Headache, hypertension, rhinitis, Raynaud phenomenon, and acrocyanosis were more frequent with sutimlimab vs placebo, with a difference of ≥3 patients between groups. Three sutimlimab patients discontinued owing to adverse events; no placebo patients discontinued. These data demonstrate that sutimlimab has potential to be an important advancement in the treatment of CAD. This trial was registered at www.clinicaltrials.gov as #NCT03347422.
Introduction
Cold agglutinin disease (CAD) is a rare type of autoimmune hemolytic anemia characterized by chronic hemolysis that is entirely mediated by activation of the classical complement pathway.1-3 CAD is a clonal, low-grade lymphoproliferative disorder that can be detected in blood or marrow in patients with no clinical or radiologic evidence of malignant conditions.4-6 CAD is distinguished from cold agglutinin syndrome, which is transient and secondary to infections, overt malignant, or autoimmune conditions.6,7 In CAD, immunoglobulin M (IgM) autoantibodies (cold agglutinins) preferentially bind to the “I” antigen on erythrocytes at temperatures ≤37°C and may result in erythrocyte agglutination.1,8-10 The antigen-IgM antibody complex, a potent trigger of the classical complement pathway, binds to the C1 complement complex, resulting in activation of C1s (a C1 complex serine protease), which activates C2 and C4, and in turn generates C3 convertase, resulting in cleavage of C3 into C3a and C3b.1,11 C3b deposition on erythrocytes results in extravascular hemolysis in the liver, the predominant mechanism of erythrocyte destruction in CAD.12-14 Although hemolysis is primarily driven by classical complement and extravascular hemolysis in CAD, severe disease may see further, terminal complement activation, with increased C3b deposition contributing to C5 convertase formation, membrane attack complex production, and intravascular hemolysis. However, this occurs to a much lesser extent than in paroxysmal nocturnal hemoglobinuria, because of intact CD55- and CD59-mediated regulation in CAD.4,15,16 Recognition of the mechanism of hemolytic anemia identified components of the classical complement pathway as a promising therapeutic target in CAD.16-18
Patients with CAD may experience complement-mediated symptoms, including chronic hemolysis resulting in anemia, profound fatigue, and jaundice; non-complement-mediated symptoms include transient, cold-induced agglutination-mediated circulatory symptoms, such as acrocyanosis and Raynaud phenomenon.4 Patients with CAD also have an increased risk of thromboembolism and early mortality.19,20 As with other causes of anemia,21-24 patients with CAD are more likely to have reduced quality of life, depression, and anxiety.25
Prior to the recent approval of sutimlimab in the United States (Sanofi 2022), there were no approved therapies for CAD.26 Supportive care with cold avoidance alone is often inadequate and does not address complement-mediated symptoms, as chronic hemolytic anemia persists year-round.2,3 Rituximab broadly depletes B cells and induces partial responses in ∼50% of patients after a median delay of 1.5 months, and relapses usually occur within 1 year.27-31 Addition of cytotoxic agents, such as fludarabine or bendamustine, to rituximab is associated with higher response rates but is accompanied by more serious toxic effects, including severe neutropenia.32,33 Eculizumab, a C5 inhibitor, reduces the need for transfusion and reduces lactate dehydrogenase (LDH) levels but elicits only a modest increase in hemoglobin (Hb) levels in patients with CAD because it does not inhibit extravascular hemolysis.34 Despite not being recommended because of poor response rates and often requiring unacceptably high doses, corticosteroids are often used in CAD management.2,35 Other approaches such as azathioprine, cyclophosphamide, and splenectomy are not effective in CAD and are also not recommended.6,36
Consequently, there is a substantial unmet need for an approved, noncytotoxic pharmacotherapy with faster onset of action, higher response rates, longer duration of response, and a favorable safety and tolerability profile.8,37 Sutimlimab (BIVV009; TNT009) is a first-in-class, humanized, monoclonal antibody designed to target C1s.38,39 By selectively inhibiting the classical complement pathway at C1s, sutimlimab leaves the immune functions of the lectin and alternative complement pathways intact.18,39-41 In the pivotal, phase 3, open-label, single-arm CARDINAL study, sutimlimab for 26 weeks had a rapid and sustained therapeutic effect in patients with CAD with a recent history of blood transfusion.38 Sutimlimab increased mean Hb levels, normalized mean bilirubin levels, and reduced transfusions.38 Moreover, sutimlimab-treated patients experienced clinically meaningful reductions in fatigue as early as week 1.38 This article presents findings from CADENZA, the first randomized, placebo-controlled phase 3 trial investigating the efficacy and safety of sutimlimab vs placebo in patients with CAD without a history of recent blood transfusion.
Methods
Trial overview
This was a 26-week, phase 3, randomized, placebo-controlled, double-blind trial (part A; completed), with an open-label extension period (part B; ongoing) in which patients continue to receive sutimlimab for a minimum of 1 year after the last patient completed part A. Only the results of part A are reported here, which was conducted between March 6, 2018 and September 29, 2020 at 53 sites across 14 countries (Australia, Austria, Belgium, Canada, France, Germany, Israel, Italy, Japan, The Netherlands, Norway, Spain, United Kingdom, and United States) in accordance with the Declaration of Helsinki protocol and the International Council for Harmonisation guidelines for Good Clinical Practice. All patients provided written informed consent for study participation. Sanofi Biostatistics analyzed the data.
Patients
At screening, adult patients with confirmed CAD diagnosis, Hb ≤ 10.0 g/dL, bilirubin level above the normal reference range, and ferritin level above the lower limit of normal were eligible. A confirmed diagnosis of CAD was defined as the presence of chronic hemolysis, positive polyspecific direct antiglobulin test, monospecific-direct antiglobulin test strongly positive for C3d, IgG-direct antiglobulin test ≤1+, a cold agglutinin titer ≥64 at 4°C, and no evidence of overt malignant disease. Patients were required to have symptomatic disease within 3 months of screening, defined as one or more of the following: symptomatic anemia, acrocyanosis, Raynaud phenomenon, hemoglobinuria, disabling circulatory symptoms, and/or major adverse vascular event, including thrombosis. Patient exclusions included: cold agglutinin syndrome secondary to infection, rheumatologic disease, or active hematologic malignancy; a history of blood transfusion within 6 months of screening or a history of >1 blood transfusion within 12 months, diagnosis of systemic lupus erythematosus or other autoimmune disorders; erythropoietin deficiency; any clinically relevant infection; recent rituximab (within 3 months)/rituximab combination therapies (within 6 months); or concurrent treatment with corticosteroids other than a stable dose equivalent to 10 mg/d prednisone for the 3 months prior. A full list of study inclusion and exclusion criteria are provided in the supplemental appendix, available on the Blood Web site.
Eligible patients were randomized 1:1 to receive 26 weeks of either sutimlimab or placebo by a permuted block randomization (block size fixed at 4) using Pharm-Olam’s Interactive Web Response System. Sutimlimab (6.5 g if body weight was <75 kg; 7.5 g if body weight was ≥75 kg) or placebo was administered by IV infusion (500 mL) over 60 minutes on day 0, day 7, and every 14 days thereafter through week 25. Patients were to receive red blood cell transfusions if they were symptomatic with Hb < 9 g/dL or asymptomatic with Hb < 7 g/dL.
Study endpoints
The primary efficacy endpoint was a composite of Hb increase from baseline of ≥1.5 g/dL at the treatment assessment timepoint (mean value from weeks 23, 25, and 26), absence of blood transfusions from week 5 to week 26, and avoidance of protocol-prohibited CAD medications from week 5 to week 26. In addition, the proportion of patients who met each of the 3 response criteria, and number of blood transfusions by study period (ie, before week 5, and between week 5 and week 26) and by treatment arm, were summarized.
Key secondary endpoints were mean change from baseline in Hb level at treatment assessment timepoint and in Functional Assessment of Chronic Illness Therapy (FACIT)-Fatigue Scale at treatment assessment timepoint (FACIT-Fatigue scores range from 0 to 52, with a higher score indicating less fatigue; an increase from baseline of ≥5 points was considered clinically significant42). Additional secondary endpoints were mean change from baseline at treatment assessment timepoint for total bilirubin level (excluding patients with either a positive Gilbert syndrome genetic test [UGT1A1 gene] or no test result recorded during screening), LDH level, haptoglobin level, and reticulocytes, and number of patients achieving mean increases of ≥1 g/dL, ≥1.5 g/dL, ≥2 g/dL, and ≥3 g/dL in Hb levels. Pharmacodynamic endpoints included changes in classical complement pathway activity (assessed using the Wieslab Complement System Classical Pathway assay [Svar Life Science, Malmö, Sweden]), complement component C4 (first soluble cleavable substrate of C1s; quantified by turbidimetry using the ADVIA 1800 Automated Chemistry analyzer with kits and reagents supplied by Siemens), C1q concentration (determined in immunoenzymatic assays with the following antibodies: capture antibody was mouse anti-C1q monoclonal antibody, clone 3R9/2 [Bio-Rad]; detection antibody was goat anti-C1q polyclonal antibody [Quidel]; secondary detection antibody was mouse anti-goat IgG (H+L)-HRP [Southern Biotech]), and hemolytic activity of the classical complement pathway (CH50) up to week 26.
Treatment-emergent adverse events (TEAE), treatment-emergent serious adverse events (TESAE), changes in autoimmune disease panel parameters, and other clinical laboratory evaluations were recorded up to week 26. Adverse events were classified according to the Medical Dictionary for Regulatory Activities, version 23.0.
Statistical analyses
A sample size of ∼40 patients (20 patients per arm) was chosen to provide statistical power >85% to detect a treatment difference of 50% improvement in response rate between sutimlimab and placebo for the composite primary endpoint, which is deemed clinically relevant. All efficacy and safety analyses were conducted using the full analysis set and safety analysis set, respectively; both sets comprised all randomized patients who received at least 1 dose of sutimlimab or placebo. To detect a treatment difference for the composite primary endpoint, the pooled 2-sided P value based on a stratified Cochran-Mantel-Haenszel test had to be <.05. Sequential closed testing was performed on key secondary endpoints with α of 0.05 for each test to protect the type I error. Tests were performed using hypothetical estimand for sutimlimab vs placebo in the following order: mean change from baseline in Hb at the treatment assessment timepoint, followed by mean change from baseline in FACIT-Fatigue score at treatment assessment timepoint. Pharmacodynamic analyses were conducted for all patients who had ≥1 dose of sutimlimab and ≥1 evaluable pharmacodynamic sample. Statistical analysis was performed using SAS version 9.4 or higher (SAS Institute Inc, Cary, NC).
Results
Baseline demographics and disease characteristics
Sixty-six patients were screened, and 42 patients were randomized to receive sutimlimab (n = 22) or placebo (n = 20) (supplemental Figure 1). Nineteen (86.4%) patients and 20 (100%) patients in the sutimlimab and placebo arm, respectively, completed part A and continued into part B. Three patients (13.6%) from the sutimlimab and none from the placebo arm discontinued part A early, owing to TEAEs. One patient (4.5%) on sutimlimab and 2 patients (10%) on placebo missed 1 or more treatment visits because of limitations imposed by the COVID-19 pandemic.
Baseline patient demographics and disease characteristics were consistent with a CAD patient population (Table 1). Most patients were female (n = 33, 78.6%), with median (range) age of 66.0 (46-88) years. At baseline, mean (standard deviation [SD]) Hb was 9.2 g/dL (1.1) and 9.3 g/dL (1.0), and mean (SD) FACIT-Fatigue score was 31.7 (12.8) and 33.0 (10.9), in the sutimlimab and placebo arms, respectively. Mean (SD) baseline bilirubin, excluding 4 patients with confirmed or undetermined Gilbert syndrome (2 patients in each group), was 41 (27) µmol/L and 36 (12) µmol/L in the sutimlimab and placebo arms, respectively. Nine patients (40.9%) in the sutimlimab group vs 4 patients (20.0%) in the placebo group had acrocyanosis, and 5 patients (22.7%) vs 3 patients (15.0%), respectively, experienced Raynaud phenomenon. Three patients in the sutimlimab group and none in the placebo group had disabling circulatory symptoms. Mean (SD) IgM load was 5.7 g/L (7.8) in the sutimlimab group and 2.7 g/L (2.0) in the placebo group (norm: 0.5-3.0 g/L). No patients had received a blood transfusion within 6 months of screening; 3 patients (13.6%) had each received a single transfusion within the previous 12 months. Thirty-one (73.8%) patients had received ≥1 CAD therapy within the previous 5 years (mostly corticosteroids and rituximab), and 5 (11.9%) patients were hospitalized for disease-related reasons within the previous 2 years.
Primary endpoint
Sixteen patients (72.7%; 95% confidence interval [CI]: 49.8, 89.3) treated with sutimlimab vs 3 patients (15.0%; 95% CI: 3.2, 37.9) who received placebo met the prespecified criteria for the composite primary endpoint and achieved the protocol-defined responder criteria (Figure 1). Patients who received 26 weeks of sutimlimab treatment had significantly greater odds of achieving the response criteria defined in the composite primary endpoint than patients receiving placebo (odds ratio [OR] 15.9; 95% CI: 2.9, 88.0; P < .001). At treatment assessment timepoint, 16 patients (72.7%) treated with sutimlimab vs 3 patients (15.0%) with placebo had an increase in Hb levels ≥1.5 g/dL from baseline. All 16 (72.7%) sutimlimab-treated patients had increased Hb levels ≥2.0 g/dL from baseline, compared with 2 (10.0%) patients on placebo (supplemental Table 1). Between weeks 5 and 26, 18 patients (81.8%) in the sutimlimab arm and 16 patients (80.0%) in the placebo arm did not receive blood transfusions. Most patients did not require use of protocol-prohibited CAD medication from week 5 to week 26 (sutimlimab n = 19, 86.4%; placebo n = 20, 100%).

Effect of sutimlimab on a composite primary endpoint comprising Hb levels, transfusions, and need for CAD medications in patients with CAD. For the composite primary endpoint, sutimlimab was compared with placebo using the Cochran-Mantel-Haenszel method, stratified by baseline Hb (< median vs ≥ median) and geographic region (Asia/Other, North America, and Europe). Hb increase from baseline of ≥1.5 g/dL was analyzed at the treatment assessment timepoint, defined as the mean average of weeks 23, 25, and 26. Requirements for transfusion included Hb < 9 g/dL and patient symptomatic or Hb < 7 g/dL and patient asymptomatic. One patient in the sutimlimab arm discontinued treatment prematurely owing to an adverse event (increased blood IgM) and started rituximab treatment during the 9-week posttreatment follow-up period; 2 patients in the sutimlimab arm discontinued prior to week 23, and their statuses were therefore “unknown” for this analysis. BL, baseline.
Effect of sutimlimab on a composite primary endpoint comprising Hb levels, transfusions, and need for CAD medications in patients with CAD. For the composite primary endpoint, sutimlimab was compared with placebo using the Cochran-Mantel-Haenszel method, stratified by baseline Hb (< median vs ≥ median) and geographic region (Asia/Other, North America, and Europe). Hb increase from baseline of ≥1.5 g/dL was analyzed at the treatment assessment timepoint, defined as the mean average of weeks 23, 25, and 26. Requirements for transfusion included Hb < 9 g/dL and patient symptomatic or Hb < 7 g/dL and patient asymptomatic. One patient in the sutimlimab arm discontinued treatment prematurely owing to an adverse event (increased blood IgM) and started rituximab treatment during the 9-week posttreatment follow-up period; 2 patients in the sutimlimab arm discontinued prior to week 23, and their statuses were therefore “unknown” for this analysis. BL, baseline.
Three patients in the sutimlimab arm did not meet the response criteria (supplemental Table 2). The patients did not achieve the required ≥1.5 g/dL increase in Hb from baseline to treatment assessment timepoint, and 1 patient received a blood transfusion. Two of these patients had an increase in Hb ≥ 1.5 g/dL over baseline on at least 1 occasion that was not associated with a blood transfusion (supplemental Figure 2). Baseline characteristics between the 2 groups are shown in supplemental Table 3. An additional 3 patients in the sutimlimab arm were excluded from the primary endpoint because they discontinued early due to TEAEs, including 1 patient who received a prohibited CAD medication. Results obtained in the 3 sensitivity analyses were consistent with the primary efficacy endpoint analysis (supplemental Table 4).
Anemia
Sutimlimab treatment resulted in rapid (within 3 weeks) and sustained increase in Hb levels, whereas no meaningful change was observed with placebo (Figure 2). Following initiation of sutimlimab, mean (standard error [SE]) Hb increased from baseline by 1.2 g/dL (0.2) at week 1, 2.0 g/dL (0.2) at week 3, and 2.7 g/dL (0.3) at treatment assessment timepoint. The proportion of patients achieving ≥1.5 g/dL increase in Hb from baseline per visit is shown in supplemental Figure 3. Overall, with sutimlimab treatment, the mean Hb level was maintained ≥11 g/dL from week 3 to treatment assessment timepoint. Fourteen (73.7%) patients in the sutimlimab arm and 4 (20.0%) patients in the placebo arm achieved Hb level ≥11 g/dL, at treatment assessment timepoint.

Effect of sutimlimab on Hb levels from baseline to week 26. Sutimlimab treatment resulted in rapid and sustained increase in Hb levels. B, baseline.
Effect of sutimlimab on Hb levels from baseline to week 26. Sutimlimab treatment resulted in rapid and sustained increase in Hb levels. B, baseline.
At treatment assessment timepoint, the least-squares (LS) mean increase from baseline in Hb was 2.66 g/dL (95% CI: 2.0, 3.22) and 0.09 g/dL (95% CI: −0.50, 0.68) in the sutimlimab and placebo arms, respectively. The LS mean difference in Hb increase between the sutimlimab and placebo arms was 2.56 g/dL (95% CI: 1.75, 3.38; P < .001 between treatments).
Hemolysis
Sutimlimab treatment led to rapid (within 1-3 weeks) control of hemolysis that was sustained throughout the study. Mean total bilirubin normalized within 1 to 3 weeks of sutimlimab and was maintained at levels below the upper limit of normal to week 26 (Figure 3A). Fifteen (88.2%) patients in the sutimlimab arm and 4 (22.2%) patients in the placebo arm achieved normal total bilirubin levels at treatment assessment timepoint. Mean (SE) change from baseline in total bilirubin at treatment assessment timepoint (excluding patients with positive/unknown Gilbert syndrome test result) was −22.1 µmol/L (2.5) in the sutimlimab arm and −1.8 µmol/L (3.3) in the placebo arm.

Effect of sutimlimab on markers of hemolysis from baseline to week 26. (A) Analysis of the change from baseline in bilirubin excluded patients with either a positive Gilbert syndrome genetic test or no test result. Overall, 21 of 22 patients in the sutimlimab arm and 18 of 20 patients in the placebo arm consented to receive Gilbert syndrome testing. Of these, no patient had a positive result, and 1 patient had unknown result and was excluded from the analyses. The normal range for bilirubin was defined as 5.1 to 20.5 µmol/L. (B) Normalization of haptoglobin was defined as greater than the level of haptoglobin detection. (C) Parameters for reticulocyte normalization were not defined for this study. (D) The normal range for LDH was defined as 120 to 246 U/L. B, baseline.
Effect of sutimlimab on markers of hemolysis from baseline to week 26. (A) Analysis of the change from baseline in bilirubin excluded patients with either a positive Gilbert syndrome genetic test or no test result. Overall, 21 of 22 patients in the sutimlimab arm and 18 of 20 patients in the placebo arm consented to receive Gilbert syndrome testing. Of these, no patient had a positive result, and 1 patient had unknown result and was excluded from the analyses. The normal range for bilirubin was defined as 5.1 to 20.5 µmol/L. (B) Normalization of haptoglobin was defined as greater than the level of haptoglobin detection. (C) Parameters for reticulocyte normalization were not defined for this study. (D) The normal range for LDH was defined as 120 to 246 U/L. B, baseline.
Similar results were obtained for additional markers of hemolysis (Figure 3B-D). Eleven (57.9%) patients in the sutimlimab arm and 6 (30.0%) patients in the placebo arm achieved normalized LDH levels at treatment assessment timepoint. A decrease in reticulocyte count coincided with the increased Hb level in sutimlimab-treated patients; reductions in reticulocyte count were not observed in placebo-treated patients. In the sutimlimab arm, an increase in the haptoglobin level corresponded to a decrease in the bilirubin level, whereas no change occurred in the placebo arm. Twelve (63.2%) patients in the sutimlimab arm and 3 (15.0%) patients in the placebo arm achieved normalized haptoglobin levels at treatment assessment timepoint.
Fatigue
Sutimlimab treatment led to rapid (by week 1) and sustained improvements in FACIT-Fatigue scores (Figure 4). Mean FACIT-Fatigue score had improved by ∼5 points by week 1 for sutimlimab-treated patients, which was clinically meaningful, vs no change for placebo-treated patients (mean change −0.1). LS mean change in FACIT-Fatigue score from baseline at treatment assessment timepoint was 10.8 points (95% CI: 7.45, 14.22) in the sutimlimab arm and 1.9 points (95% CI: −1.65, 5.46) in the placebo arm. The LS mean score difference between the sutimlimab and placebo arms at treatment assessment timepoint was 8.9 points (95% CI: 4.0, 13.85; P < .001 between treatments).
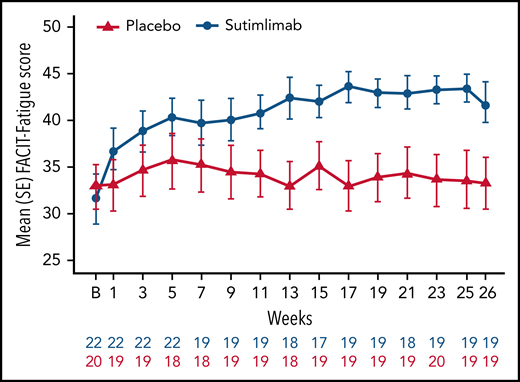
Effect of sutimlimab on FACIT-Fatigue Scale scores from baseline to week 26. FACIT-Fatigue Scale scores range from 0 to 52, with a higher score indicating less fatigue. In CAD, a change of 5 is estimated to be a clinically important change.42 B, baseline.
Effect of sutimlimab on FACIT-Fatigue Scale scores from baseline to week 26. FACIT-Fatigue Scale scores range from 0 to 52, with a higher score indicating less fatigue. In CAD, a change of 5 is estimated to be a clinically important change.42 B, baseline.
Pharmacodynamics
Normal serum ranges for the Wieslab Complement System Classical Pathway assay, C4, and CH50 are 69%-129%, 0.18-0.45 g/L, and 31.6-57.6 U/mL, respectively. Below-normal ranges are expected in CAD and are consistent with this disease. Sutimlimab treatment resulted in near-complete inhibition of classical complement pathway activity, which was sustained throughout the treatment period (Figure 5A). Mean (SE) predose classical complement pathway activity at baseline was 22.4% (4.2) in the sutimlimab arm and 32.8% (6.4) in the placebo arm. After the first dose of sutimlimab, at week 1, mean (SE) classical complement pathway activity decreased to 2.3% (0.6) in the sutimlimab arm, whereas the mean classical complement pathway activity remained similar to baseline in the placebo arm (35.6% [5.8]). Sutimlimab-induced reductions in mean (SE) classical complement pathway activity were maintained to week 26 (sutimlimab, 3.2% [0.5] vs placebo, 29.3% [7.0]).
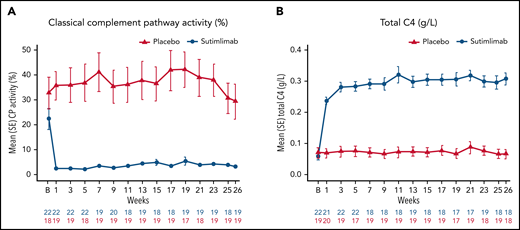
Effect of sutimlimab on classical complement pathway activity. (A) In the Wieslab Complement System Classical Pathway assay, the normal range in serum is 69% to 129%. (B) The International System of Units reference range for serum C4 is 0.18 to 0.45 g/L. Baseline classical complement pathway activity and total C4 levels below the normal ranges are expected in CAD and are consistent with the nature of this disease. B, baseline; C4, complement component 4; CP, classical complement pathway.
Effect of sutimlimab on classical complement pathway activity. (A) In the Wieslab Complement System Classical Pathway assay, the normal range in serum is 69% to 129%. (B) The International System of Units reference range for serum C4 is 0.18 to 0.45 g/L. Baseline classical complement pathway activity and total C4 levels below the normal ranges are expected in CAD and are consistent with the nature of this disease. B, baseline; C4, complement component 4; CP, classical complement pathway.
Mean (SE) predose total C4 levels in the sutimlimab arm were 0.06 g/L (0.01), 0.24 g/L (0.01), and 0.31 g/L (0.02) at baseline, week 1, and week 26, respectively (Figure 5B). By contrast, mean (SE) predose C4 level in the placebo arm was unchanged from baseline (0.07 g/L [0.02]), through week 1 (0.07 g/L [0.02]), and week 26 (0.07 g/L [0.02]). At baseline, mean (SE) CH50 levels were 27.0 (3.3) U/mL for the sutimlimab arm and 20.8 (20) U/mL for placebo. After the first dose of sutimlimab and throughout the treatment period, the mean value of CH50 for sutimlimab was below limit of quantification, whereas the mean (SE) values for the placebo group ranged from 15.6 (4.3) to 23.5 (5) U/mL. C1q levels generally remained unchanged through the treatment period (supplemental Figure 4).
Safety
Overall, 21 patients (96%) in the sutimlimab arm experienced a total of 146 TEAEs, and 20 patients (100%) in the placebo arm experienced a total of 90 TEAEs (Table 2). The majority of TEAEs were considered unrelated to the study treatment by the investigator. Eight patients (36%) experienced 28 TEAEs that were considered related to sutimlimab treatment, and 4 patients (20%) experienced 7 TEAEs that were considered related to placebo administration by the investigator, with no specific trend observed in either group. Headache (22.7% vs 10.0%), hypertension (22.7% vs 0%), rhinitis (18.2% vs 0%), Raynaud phenomenon (18.2% vs 0%), and acrocyanosis (13.6% vs 0%) were reported more frequently in sutimlimab-treated patients compared with placebo, with a difference of ≥3 patients between groups (supplemental Table 5).
In the sutimlimab arm, 3 patients (14%) experienced a total of 4 TESAEs: febrile infection and increased blood IgM (n = 1), Raynaud phenomenon (n = 1), and cerebral venous thrombosis (n = 1). One patient with elevated IgM levels at screening and baseline experienced 2 serious events: 1 TESAE of febrile infection and 1 TESAE of increased blood IgM. A nonserious TEAE of increased blood IgM was reported on day 36. The IgM level was noted to increase on day 42, and this event was then reported as a TESAE. There was no clinical evidence for hyperviscosity syndrome. The patient was diagnosed with low-grade B-cell lymphoma. Sutimlimab was permanently discontinued, and the patient received rituximab as treatment for elevated IgM, which resolved on day 169. Both events were assessed by the investigator as not related to sutimlimab. One patient with a history of Raynaud phenomenon had a TESAE of Raynaud phenomenon requiring hospitalization, which was considered not related to sutimlimab treatment by the investigator. One elderly patient with history of diabetes had a severe cerebral venous thrombosis on day 86. Sutimlimab was temporarily interrupted, and the TESAE resolved on day 88 (without sequalae) after treatment with antithrombotic agents. This TESAE was assessed by the investigator as possibly related to sutimlimab. In the placebo arm, 1 patient (5%) experienced 3 TESAEs: 2 TESAEs of anemia that occurred on days 21 and 63 and resolved on days 22 and 66, respectively, and 1 TESAE of vascular device infection on day 64 that resolved on day 66. These were all assessed as not related to placebo by the investigator.
Three patients (14%) in the sutimlimab arm discontinued the study drug prematurely because of TEAEs and TESAEs. One patient had multiple TEAEs of acrocyanosis with concurrent Raynaud phenomenon and was later diagnosed with lymphoproliferative disease progression. One patient discontinued because of 1 TESAE of blood IgM increase and was later diagnosed with low-grade B-cell lymphoma, and the third patient had an infusion-related reaction (pain in lumbar spine and both legs during sutimlimab infusion) assessed as possibly related to sutimlimab by the investigator.
Eighteen TEAEs of infection were reported by 10 patients (45%) in the sutimlimab arm, and 19 TEAEs of infection were reported in 10 patients (50%) in the placebo arm. Two serious infections were reported, one in each treatment arm. One patient in the sutimlimab group experienced a serious event of “febrile infection of unknown origin” on day 2 that required hospitalization and resolved on day 23. One patient in the placebo arm had a vascular device infection caused by Bacillus cereus. Neither serious infection led to treatment discontinuation. There were no meningococcal infections. A single patient in the study (placebo arm) experienced a nonserious event of suspected COVID-19 infection. No development of systemic lupus erythematosus or other autoimmune disorders or worsening of preexisting autoimmune disorders were reported. No serious hypersensitivity reactions and/or anaphylaxis were reported, and no deaths occurred during the study.
There are no available data on COVID-19 vaccination from this study.
Clinical laboratory parameters
Median (range) D-dimer levels were 401 (190, 2194) μg/L fibrinogen equivalent units at baseline and 271 (190, 8537) μg/L fibrinogen equivalent units at week 26 in the sutimlimab arm (n = 21), compared with median 392 (190, 1837) μg/L fibrinogen equivalent units at baseline and 475 (190, 3845) μg/L fibrinogen equivalent units in the placebo arm (n = 20). In 1 subject in the sutimlimab arm, D-dimer increased from 391 μg/L fibrinogen equivalent units at baseline to 8537 μg/L fibrinogen equivalent units at week 26. No clinical correlates (including thromboembolic events) were associated with this increase.
Discussion
CADENZA is the first placebo-controlled trial of sutimlimab in CAD and strengthens the results from the previous single-arm open-label CARDINAL study of patients with CAD and recent transfusion history.38 Sutimlimab led to normalization of mean Hb levels and markers of hemolysis, and clinically meaningful improvements in fatigue in patients with CAD without a history of recent blood transfusion. Response to sutimlimab was rapid (within 1-3 weeks) and sustained over 26 weeks. Mean Hb levels were maintained at >11 g/dL with sutimlimab from week 3 through to study end. Mean total bilirubin (a marker of hemolysis) was normalized within 1 to 3 weeks with sutimlimab and maintained at levels below the upper limit of normal to week 26. Conversely, placebo had no significant effect on these endpoints.
At baseline, FACIT-Fatigue scores for patients with CAD in CADENZA were similar to scores seen in patients with serious chronic conditions, including rheumatoid arthritis,43 advanced cancer-related anemia,44 and paroxysmal nocturnal hemoglobinuria.45,46 A clinically meaningful change in FACIT-Fatigue in autoimmune or oncologic diseases has been shown to be in the range of 3 to 10.47-49 In CAD, a change in FACIT-Fatigue of ≥5 points is estimated to be a clinically important change.42 In this trial, treatment with sutimlimab resulted in significantly improved LS mean FACIT-Fatigue score compared with placebo, as well as a >10-point mean increase in FACIT-Fatigue score compared with baseline in the sutimlimab group at treatment assessment timepoint, demonstrating improved quality of life. The connection between inflammation and fatigue has been explored in a study showing that patients with CAD have elevations in both the proinflammatory cytokine interleukin (IL)-6 and the regulatory cytokine IL-10. Treatment of patients with CAD with sutimlimab reduced levels of both inflammatory markers (IL-6, IL-10) with concurrent improvement in patients’ fatigue scores, demonstrating the influence of complement inhibition on inflammation in CAD and suggesting a role for complement-mediated inhibition in fatigue.50
The observed efficacy results in CADENZA correlated with near-complete inhibition of classical complement pathway activity from week 1 through end of treatment in the sutimlimab group. Classical complement pathway activity was below the normal range at baseline, which is consistent with the disease nature (continuous complement activation leading to consumption of complement components) and corroborated by the low levels of total C4 and CH50 observed at baseline. As C4 is the first substrate cleaved following activation of C1s, C4 levels are low in patients with CAD, owing to cold agglutinin-mediated classical complement pathway activation and continuous consumption. At week 26, mean classical complement pathway activity was almost completely inhibited in the sutimlimab arm. Blockade of C1s by sutimlimab increased circulating levels of C4 several-fold in patients, providing an in vivo marker of sutimlimab activity.
Sutimlimab treatment was generally well tolerated. The type and frequency of TEAEs were consistent with an elderly and medically complex patient population. Headache, hypertension, rhinitis, Raynaud phenomenon, and acrocyanosis were more frequent with sutimlimab vs placebo, with a difference of ≥3 patients between groups. No deaths were reported, and there were no reports of meningococcal infections. Serious infections were reported without leading to treatment discontinuation. No development of systemic lupus erythematosus or serious hypersensitivity reactions and/or anaphylaxis were reported in either group. A limitation of this study is that patient numbers were low but consistent with a rare disease with clinical heterogeneity that may have contributed to some baseline imbalances (higher female:male ratio). Observed baseline imbalances, including disabling circulatory symptoms, acrocyanosis, Raynaud phenomenon, and IgM levels (Table 1), may have impacted the results observed. Of note, the baseline imbalance in incidence of CAD symptoms was less distinct at week 26 (supplemental Table 6). Real-world evidence studies may help further characterize the effectiveness and safety of sutimlimab in CAD.
In conclusion, CADENZA phase 3 data support the targeted inhibition of C1s as an effective and well-tolerated treatment for patients with CAD. Statistically significant and clinically meaningful differences were demonstrated between sutimlimab and placebo, further supporting the efficacy of sutimlimab. Sutimlimab has a novel and targeted mechanism of action that specifically addresses the underlying cause of chronic hemolytic anemia in CAD. These data show that targeting the classical complement pathway at C1s represents a new, effective therapeutic approach for CAD management, independent of transfusion status, with treatment responses as early as week 1 and a favorable tolerability profile.
Acknowledgments
The authors thank the investigators, health care providers, research staff, and patients who participated in the CADENZA study.
This study was funded by Sanofi (Waltham, MA). Support with statistical analyses was provided by Braydon Schaible and Jennifer Wang (Study Lead Statisticians, Sanofi).
Medical writing and editing support was provided by Lisa Buttle and Katharina Schleicher of Lucid Group, funded by Sanofi.
Authorship
Contribution: A.R., S.B., W.B., B.J., I.C.W., M.Y., J.M.I.V., N.W., X.J., M.W., F.S., and C.M.B. contributed substantially to conception and design, data acquisition, and data analysis or interpretation of data for the work related to this document; S.D., M.M., J.-i.N., and M.L. contributed substantially to data acquisition, and data analysis or interpretation of data for the work related to this document; M.S. contributed substantially to data analysis or interpretation of data for the work related to this document; P.P. and D.S.V. contributed substantially to conception and design, and data analysis or interpretation of data for the work related to this document; and all authors had access to primary clinical trial data, had full editorial control of the manuscript, and provided their final approval of all content.
Conflict-of-interest disclosure: A.R. has received research support from Roche, received honoraria, and provided consultancy to Alexion Pharmaceuticals, Inc, Apellis Pharmaceuticals, Novartis, Roche, Bioverativ, a Sanofi company, and Sanofi. S.B. has received research funding from Mundipharma; lecture honoraria from Apellis Pharmaceuticals, Alexion Pharmaceuticals, Inc, Bioverativ, Janssen-Cilag, and Sanofi; and provided consultancy to Apellis Pharmaceuticals, Bioverativ, Sanofi, Sobi, and True North Therapeutics. W.B. has received research support from Alexion Pharmaceuticals, Inc and Novartis; participated in advisory boards for Agios, Alexion Pharmaceuticals, Inc, Bioverativ, and Incyte; and has been an invited speaker for Alexion Pharmaceuticals, Inc, and Novartis. S.D. has received grant funding, honoraria, and/or speakers fees from Janssen, BeiGene, and Sanofi. B.J. has received reimbursement for travel costs for scientific presentations and consultancy to True North Therapeutics, Bioverativ, and Sanofi. M.M. has provided consultancy to Alexion Pharmaceuticals, Inc, Rigel, and Bioverativ. J.-i.N. has received honoraria and provided consultancy to Sanofi, Alexion, Chugai, Roche, Novartis, Apellis, and Biocryst. J.M.I.V. has received research funding from BeiGene and Takeda, travel support from Cellgene, and is an international advisory board member for Sanofi. M.S., N.W., X.J., F.S., M.L., and M.W. are employees of Sanofi and may hold shares and/or stock options in the company. P.P. was an employee of Sanofi at the time of the study. D.S.V. is an employee of IQVIA and provided safety data analysis and support under contract to Sanofi. C.M.B. has received honoraria and/or research funding from Alexion Pharmaceuticals, Inc, Bioverativ, Cellphire, Incyte, Rigel, and Sanofi Genzyme. The remaining authors declare no competing financial interests.
Correspondence: Alexander Röth, Department of Hematology and Stem Cell Transplantation, West German Cancer Center, University Hospital Essen, University of Duisburg-Essen, Hufelandstrasse 55, 45147 Essen, Germany; e-mail: alexander.roeth@uk-essen.de.
Presented in abstract form at the European Hematology Association annual meeting, 9-17 June 2021 (virtual meeting).
Qualified researchers may request access to anonymized patient-level data and related study documents (redacted to protect participants’ privacy). Further details can be found at https://www.vivli.org/.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.