

Properdin is a positive regulator of alternative pathway (AP) complement. The current understanding of properdin function is that it facilitates AP complement activation by stabilizing the C3 convertase C3bBb. Properdin-deficient patients are susceptible to lethal meningococcal infection, but the mechanism of this selective predisposition is not fully understood. By gene targeting in the mouse, we show here that properdin is essential for AP complement activation induced by bacterial lipopolysacharride (LPS) and lipooligosacharride (LOS) and other, but not all, AP complement activators. LPS- and LOS-induced AP complement activation was abolished in properdin−/− mouse serum, and properdin−/− mice were unable to clear Crry-deficient erythrocytes, which are known to be susceptible to AP complement–mediated extravascular hemolysis. In contrast, zymosan- and cobra venom factor–induced AP complement activation, and classical pathwaytriggered AP complement amplification were only partially or minimally affected in properdin−/− mice. We further show that the ability of human properdin to restore LPS-dependent AP complement activity in properdin−/− mouse serum correlated with the human properdin-binding affinity of the LPS. These results reveal a novel role of properdin in AP complement initiation and have implications for understanding the selective predisposition of properdin-deficient patients to meningococcal infection.
Introduction
The complement system provides a first line of host defense against invading pathogens. Activation of complement occurs via 3 different pathways: the classical, lectin, and alternative pathway.1 The classical pathway is initiated by antigen–antibody binding.1 The lectin pathway is triggered when mannose-binding lectins (MBL) interact with surface sugar molecules on microorganisms. Activation of both pathways leads to the assembly of the classical pathway C3 convertase C4b2a, although direct cleavage of C3 by MBL–associated serine proteases can also occur.2 The alternative pathway (AP) is a self-amplification loop driven by the AP C3 convertase, C3bBb.1 AP activation can occur secondary to classical or lectin pathway activation or be initiated independently. In the latter case, a low level spontaneous C3 “tick-over” generates the initial C3bBb, which rapidly propagates AP in the absence of adequate regulation.1 Thus, AP complement activation on nonself surfaces with no or insufficient negative regulation is considered a default process, whereas autologous cells typically avoid this outcome with the help of multiple membrane–bound and fluid phase complement inhibitory proteins.3
In contrast to the existence of numerous inhibitory proteins, the plasma protein properdin is the only known physiologic positive regulator of the complement activation cascade.4 Discovered more than 50 years ago, properdin was at first regarded as an initiator of the AP complement, acting in a manner that was analogous to antibodies of the classical pathway.5,,–8 This concept of properdin function caused controversy4,9 and was eventually abandoned, to be replaced by the currently held view that properdin facilitates AP complement activation by extending the half-life of the nascent C3bBb convertase.4,10 Of interest, a recent study has demonstrated that surface C3b–bound properdin could serve as a platform for new C3bBb assembly.11 This pointed out a more complex mechanism of action of properdin in AP complement activation and brought forward the need for further investigation on properdin function.
Structurally, properdin is composed of an N-terminal domain and 6 thrombospondin type I repeat (TSR) domains.4 Under physiologic conditions, it exists in plasma as cyclic polymers (dimers, trimers, tetramers), formed by head to tail associations of monomers.12,13 Human properdin is encoded on the short arm of the X chromosome; and its deficiency, especially when combined with C2, MBL, or immunoglobulin G2 (IgG2) deficiency,14,–16 constitutes a high penetrance risk factor for lethal Neisseria infections. The mechanism of this selective predisposition to Neisseria infections in human properdin-deficient patients is not fully understood.17
In the present study, we have generated a properdin knockout mouse and used it to assess the role of properdin in AP complement activation in vitro and in vivo. We report here the unexpected finding of activator-specific requirement of properdin in AP complement activation, and demonstrate the potential of properdin as an initiator of AP complement. These results shed new light on the mechanism of action of properdin and have implications for understanding the susceptibility of properdin-deficient patients to meningococcal infection.
Methods
Properdin gene targeting
To construct the targeting vector, we used the pND1 vector which contains neomycin (NEO) and diphtheria toxin (DT) as a positive and negative selection marker, respectively (kindly provided by Dr Glen Radice, University of Pennsylvania). This vector contains 2 LoxP sites for Cre recombinase–mediated gene excision, and the NEO was flanked by 2 FLP recombinase target (FRT) sites for potential excision by the FLPe recombinase.18 Properdin gene fragments were amplified by polymerase chain reaction (PCR) using 129/Sv mouse genomic DNA as template and with the Expand Long Template PCR System (Roche, Indianapolis, IN). For the 3′ homologous arm, a 3.5-kb gene fragment containing exons 6-9 was amplified using 5′-GAA-TTC-TTG-TAC-CAC-GTG-ATC-3′ and 5′-TCCCCATACTCAGCACTATTG-3′ as primers, cloned into the PCR 2.1 vector (Invitrogen, Carlsbad, CA), and then subcloned into the EcoRI site in pND1 (downstream of the NEO cassette, Figure 1B). For the 5′ homologous arm, 2 fragments, a 4-kb NotI-EcoRV fragment containing exons 1-2 and a 1.6-kb EcoR V-XhoI fragment containing exons 3-5, as well as incorporating a 34 bp LoxP site (5′-ATAACTTCGTATAATGTATGCTATA-CGAAGTTAT-3′), were amplified using the following primer pairs: 5′-GATATCAT-AACTTCGTATAATGTATGCTATACGAAGTTATGTTCAATCACCCACCATCC-CT-3′ and 5′-CTCGAGCATTCATCTTTGCCAGAAATC-3′; 5′-GCGGCCGCATTCC-TGGCTGTATCTGAGTC-3′ and 5′-GATATCAGGAAGAAGTGAATATACAGG-3′. These 2 pieces were cloned into the pND1 vector at NotI-XhoI sites (upstream of the Neo) in a 3-piece ligation experiment.

Generation of properdin−/− mice. (A) Schematic representation of the mouse properdin gene locus. Vertical columns symbolize exon (E) locations. Horizontal rectangle box indicates the location of cDNA probe used for ES cell screening. (B) Targeting vector. Big arrowheads represent LoxP sites and small arrowheads represent FRT sites. Neo indicates neomycin; and DT, diphtheria toxin. (C) Actual recombinant properdin gene locus. (D) Expected restriction fragment lengths of wild-type and recombinant alleles. (E) Representative Southern blot screening result of ES cells after HincII and ScaI digestion. (F) Northern blot analysis of properdin mRNA in wild-type (WT) and properdin knockout (P−/−) mouse tissues. (G) Immunodiffusion analysis of properdin in plasma. Antihuman properdin antibody was placed in the center well and mouse (10 μL) and human (5 μL) plasma or purified human properdin (0.5 μg) was placed in the peripheral wells. A precipitation line between the center and a peripheral well indicates the presence of properdin in the testing sample. (H) Western blot analysis showing the lack of properdin protein (P, 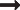 ) in properdin−/− mouse serum. The band below properdin represents goat IgG heavy chain used in immunoprecipitation. Purified human properdin (hP) was used as a positive control on the right lane. The size (in kDa) and position of molecular weight markers was shown on the left.
) in properdin−/− mouse serum. The band below properdin represents goat IgG heavy chain used in immunoprecipitation. Purified human properdin (hP) was used as a positive control on the right lane. The size (in kDa) and position of molecular weight markers was shown on the left.
Generation of properdin−/− mice. (A) Schematic representation of the mouse properdin gene locus. Vertical columns symbolize exon (E) locations. Horizontal rectangle box indicates the location of cDNA probe used for ES cell screening. (B) Targeting vector. Big arrowheads represent LoxP sites and small arrowheads represent FRT sites. Neo indicates neomycin; and DT, diphtheria toxin. (C) Actual recombinant properdin gene locus. (D) Expected restriction fragment lengths of wild-type and recombinant alleles. (E) Representative Southern blot screening result of ES cells after HincII and ScaI digestion. (F) Northern blot analysis of properdin mRNA in wild-type (WT) and properdin knockout (P−/−) mouse tissues. (G) Immunodiffusion analysis of properdin in plasma. Antihuman properdin antibody was placed in the center well and mouse (10 μL) and human (5 μL) plasma or purified human properdin (0.5 μg) was placed in the peripheral wells. A precipitation line between the center and a peripheral well indicates the presence of properdin in the testing sample. (H) Western blot analysis showing the lack of properdin protein (P, ) in properdin−/− mouse serum. The band below properdin represents goat IgG heavy chain used in immunoprecipitation. Purified human properdin (hP) was used as a positive control on the right lane. The size (in kDa) and position of molecular weight markers was shown on the left.
The targeting vector was linearized by Not I digestion before transfection. ES cells were selected with G418 (0.2 mg/mL) and positive clones were screened by Southern blot using HincII- and Sca I-digested genomic DNAs and a 513-bp probe located 3′ to the right homologous arm (Figure 1A-D). ES cell culture, vector transfection, clone selection, and chimera mouse production were carried out as described.19 For PCR genotyping, 5′-GGGTGGGATT-AGATAAATGCC-3′ (P1, NEO-specific) and 5′-CAAGGTACGGCTTTGTTACACA-3′ (P2, properdin-specific) were used for NEO detection (700 bp product), 5′-ATAACTTCGTATAATGTATGCTATACGAAGTTAT-3′, and P2 were used for LoxP detection (400 bp product). 5′-CACTGATATTGTAAGT-AGTTTGC-3′ and 5′-CTAGTGCGAAGTAGTGAT CAGG-3′ were used for FLPe transgene detection.18
Mice and reagents
Properdin-deficient (properdin−/−) mice developed, grew, and reproduced normally. Male properdin−/− mice and their WT littermates on a mixed 129/C57BL/6 background were used in all experiments. FLPe-Tg (B6;SJL-Tg(ACTFLPe)9205 Dym/J) mice were from the Jackson Laboratory (Bar Harbor, ME). C3−/−, fB−/−, and Crry−/−C3−/− mice were as described previously.20 Rabbit anti-ovalbumin (OVA) and rabbit anti-C3c antibodies were kindly provided by Dr J. Lambris (University of Pennsylvania). Zymosan A (Saccharomyces cerevisiae), S typhosa, S minnesota (S), E coli 026:B6 LPS, OVA, and HRP antimouse IgG were from Sigma-Aldrich. Human properdin was from Quidel (San Diego, CA). Anticore LPS mAb, WN1 222-5, was from Cell Sciences (Canton, MA). Goat antihuman properdin and fB antibodies were from Complement Technologies (San Diego, CA). HRP goat anti-C3 antibody was from MP Biomedicals (Solon, OH). Neisseria meningitidis LOS21 was kindly provided by Dr Sanjay Ram (University of Massachusetts, Worcester, MA). All animal experiments were approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania.
Northern blot and immunodiffusion assay
Northern blot analysis was performed as described19 using a 700-bp mouse properdin cDNA as a probe. Plasma properdin was detected by immunodiffusion assays as described in Current Protocols in Immunology (John Wiley and Sons) using antihuman properdin antibodies.
Immunoprecipitation and Western blot analysis
Mouse properdin was immunoprecipitated by incubating 200 μL pooled plasma (from 3 properdin−/− or WT littermate controls) with 10 μg of goat antihuman properdin IgG for 3 hours at 4°C. The immune complexes were then retrieved by adding 30 μL protein G agarose (Invitrogen) followed by overnight incubation. The agarose pellet was washed and boiled in sample buffer, centrifuged, and supernatant loaded onto a 10% SDS-PAGE gel under reducing condition. Proteins were transferred to PVDF membrane as previously described.22 The membrane was incubated with goat anti–human properdin IgG (5 μg/mL), followed by incubation with HRP-conjugated rabbit anti–goat IgG (1:4000; Bio-Rad Laboratories, Hercules, CA). After washing, the membrane was developed using the ECL Kit.
Detection of plate-bound LPS
To compare the plate-coating efficiency of LPS from different bacterial species (S typhosa, S minnesota, and E coli), plates were coated with diluted concentrations of LPS. After blocking with bovine serum albumin (BSA) (10 mg/mL) for 1 hour, plates were washed with phosphate-buffered saline and incubated for 1 hour with WN1 222-5 (0.5 μg/mL), a murine anticore LPS monoclonal antibody, followed by detection with HRP-anti mouse IgG (1:6000). BSA-coated wells were used as background controls.
Enzyme-linked immunosorbent assays of complement activation
Plates were coated with LPS (2 μg/well) or OVA/anti-OVA immune complex for complement activation assays as described previously.23 Diluted mouse serum (50 μL per well) was incubated on plates at 37°C for 1 hour followed by detection of plate–bound activated C3 using HRP antimouse C3 antibody (1:4000). AP activity was assayed in Mg2+-ethylene glycol tetraacetic acid (EGTA) and total or classical pathway activity was assayed in GVB2+. For reconstitution experiments, 1:10 diluted (in Mg2+-EGTA) C3−/− serum was premixed (at 1:1 ratio) with variously diluted properdin−/− serum. Alternatively, human properdin was added to properdin−/− serum (62.5 ng to 50 μL serum) or used to pretreat LPS-coated plates (62.5 ng in 25μL Mg2+-EGTA, 1 hour at 37°C, followed by washing). To deplete fB, properdin−/− serum was preincubated with antihuman fB IgG (8 μg/μL serum), followed by centrifugation to remove anti-fB/fB immune complexes.
LPS-properdin binding
In the first assay, plates were coated with different concentrations of LPS, blocked with BSA (10 mg/mL), and then incubated with purified human properdin (2.5 μg/mL in Mg2+-EGTA, 25 μL/well) at RT for 1 hour, followed by washing and detection with biotinylated antiproperdin IgG (2 μg/mL) and avidin-HRP (1:10,000). LPS–coated wells not treated with properdin were used as background controls. In the second assay, plates were coated with a fixed concentration of LPS (5 μg/well) and then incubated with increasing concentrations of purified human properdin.
Measurement of AP activation on zymosan
CVF treatment in vitro
Serum (5μL) was incubated with 0.01μg or 0.3 μg cobra-venom factor (CVF) for various lengths of time. After incubation, 0.5 μL serum was run on an 8% gel under reducing conditions and subjected to Western blot analysis as described24 using HRP-conjugated rabbit antimouse C3 antibody. C3 cleavage was quantified by densitometry scanning of activated and intact C3 α-chain.
Erythrocyte transfusion and survival assay
Sensitivity of Crry-deficient mouse erythrocytes to AP complement attack in vivo was assayed as described.19
In vivo complement activation induced by LOS or LPS
Mice were injected (intraperitoneally) with 20 mg/kg N meningitidis LOS or S typhosa LPS. Plasma levels of activated C3 were determined at 1 hour after treatment as described.26
Results
Generation of a properdin knockout (properdin−/−) mouse
The mouse properdin gene is located on the X chromosome and is composed of 9 exonss (http://www.informatics.jax.org/searches/accession_report.cgi?id = MGI:97 545; Figure 1A). Our original plan was to generate a conditional properdin gene knockout mouse so that the significance of its tissue-specific production could be studied. To achieve this goal, we constructed a targeting vector by cloning 5′ and 3′ homologous arm sequences into the pND1 vector as illustrated in Figure 1B. According to this strategy, after correct targeting the neomycin cassette (NEO) would be inserted between exons 5 and 6 of the properdin gene, and exons 3-5 would be flanked by 2 LoxP sites (Figure 1B), allowing them to be deleted by tissue-specific Cre recombinase.27 We targeted exons 3-5 for deletion because mutations in exons 4-6 of the human properdin gene are associated with properdin deficiency.15,28,–30 Targeted embryonic stem (ES) cells were selected by Southern blot analysis after HincII and Sca I digestion of genomic DNA (Figure 1C-E), using a 513 bp probe located outside the 3′ homologous arm. We obtained 7 positive ES cell clones and used 2 of them for chimeric mice production. Chimeras derived from both ES cell clones successfully transmitted the mutation through the germ line.
Subsequent analysis of the recombinant properdin gene allele, both in the mutant mice and in the 2 ES cell clones used to generate them, confirmed NEO insertion at the intended location but failed to detect the 5′ LoxP sequence (Figure 1C; analysis data not shown). The latter outcome was unexpected but most likely occurred as a result of homologous recombination in the sequence downstream (ie, exons 3-5) rather than upstream (ie, exons 1-2 and 5′ flanking region) of the 5′ LoxP site (Figure 1A,B). Nevertheless, Northern blot analysis revealed the absence of a 1.4-kb properdin mRNA species in various tissues of the mutant mice (Figure 1F). A band between 4.7 and 1.9 kb, present in both the wild-type (WT) and the mutant mouse spleens, most likely represented a nonspecific signal as real-time PCR analysis failed to detect any properdin mRNA in multiple tissues of the mutant mice (data not shown). Furthermore, immunodiffusion and Western blot analysis confirmed the lack of properdin protein in the mutant mouse plasma (Figure 1G,H). These results suggested that NEO insertion into the small intron (201 bp) between exon 5 and 6 might have unintentionally disrupted the properdin gene. To verify this conclusion, we crossed the properdin mutant mouse with the FLPe transgenic mouse. The NEO cassette in our targeting construct was flanked by 2 FRT sites, which could be recognized by the FLPe recombinase.18 As expected, expression of the FLPe recombinase eliminated NEO from the genome of properdin gene-targeted mice with corresponding recovery of properdin protein in their plasma (Figure 2). Thus, by NEO insertion into the 5th intron, we unexpectedly created a global properdin gene knockout mouse (properdin−/−).

Rescue of properdin gene knockout by NEO deletion. (A) Schematic diagram showing expected recombinant properdin gene locus after FLPe-mediated NEO deletion. (B) PCR genotyping of 7 mice derived from properdin−/− × FLPe-transgenic mouse crossing. Using LoxP or FLPe-specific primers, 2 mice (numbers 1 and 5) were identified as having recombinant properdin gene and 4 mice (numbers 1-4) were FLPe transgenic. As expected, the FLPe-negative, LoxP-positive mouse (number 5) contained NEO, whereas the FLPe-positive, LoxP-positive mouse (number 1) did not contain NEO. (C) Immunodiffusion analysis of plasma properdin showing that no properdin was present in mouse 5 (properdin−/−), whereas properdin was detected in mouse 1 (knockout rescued). Antihuman properdin antibodies were placed in the center well, and plasma samples for mice 1, 2, 5, and 6 (B) were placed in the peripheral wells.
Rescue of properdin gene knockout by NEO deletion. (A) Schematic diagram showing expected recombinant properdin gene locus after FLPe-mediated NEO deletion. (B) PCR genotyping of 7 mice derived from properdin−/− × FLPe-transgenic mouse crossing. Using LoxP or FLPe-specific primers, 2 mice (numbers 1 and 5) were identified as having recombinant properdin gene and 4 mice (numbers 1-4) were FLPe transgenic. As expected, the FLPe-negative, LoxP-positive mouse (number 5) contained NEO, whereas the FLPe-positive, LoxP-positive mouse (number 1) did not contain NEO. (C) Immunodiffusion analysis of plasma properdin showing that no properdin was present in mouse 5 (properdin−/−), whereas properdin was detected in mouse 1 (knockout rescued). Antihuman properdin antibodies were placed in the center well, and plasma samples for mice 1, 2, 5, and 6 (B) were placed in the peripheral wells.
Abrogation of LPS–induced AP complement activation in properdin−/− mouse serum
To assess AP complement activity in properdin−/− mouse serum, we used an enzyme-linked immunosorbent assay (ELISA) to measure LPS-induced complement activation in Mg2+-EGTA.23 LPS was coated onto 96-well plates and after exposure to mouse serum, the level of C3 deposition on the plates was determined. Using a broadly cross-reacting anticore LPS monoclonal antibody, we first confirmed that LPS from 3 different bacteria species, S typhosa, S minnesota (S), and E coli bound to ELISA plate with similar avidity (Figure 3A). Figure 3B-D shows that these LPS all activated AP complement in wild-type (WT) mouse serum. In contrast, the same LPS did not cause appreciable AP complement activation in properdin−/− mouse serum or in WT mouse serum treated with EDTA (ethylenediaminetetraacetic acid; negative control; Figure 3B-D). Addition of C3−/− mouse serum (as a source of murine properdin) or purified human properdin to properdin−/− mouse serum restored S typhosa LPS–induced AP complement activity to WT or higher levels (Figure 3B). Importantly, pretreatment of S typhosa LPS–coated plates with human properdin followed by washing also reconstituted AP complement activation in properdin−/− mouse serum (Figure 3B). This result suggested that purified human properdin was able to bind to S typhosa LPS with sufficient affinity and that immobilized LPS-bound properdin activated AP complement in the absence of solution properdin.

ELISAs of LPS-induced AP complement activation. (A) ELISA detection of LPS on LPS-coated plates. (B) AP complement activation by plate-bound S typhosa LPS in wild-type (WT) or properdin knockout (P−/−) mouse serum. To reconstitute AP activity in properdin−/− mouse serum, C3−/− serum or purified human properdin (hP) was premixed with properdin−/− mouse serum. Alternatively, LPS-coated plates were incubated with hP and washed (hP coat) before exposure to properdin−/− serum. Similar assays were performed with plate-bound LPS from S minnesota (S) (C) or E coli (D). (E,F) ELISAs of hP interaction with plate-bound LPS. Plates were first coated with different concentrations of LPS and then incubated with a fixed concentration of purified hP (62.5 ng/well; E). (F) Plates were first coated with a fixed concentration of LPS (5 μg/mL) and then incubated with increasing concentrations of purified hP. After washing, the amount of plate-bound properdin was detected by antiproperdin antibodies. OD, optical density.
ELISAs of LPS-induced AP complement activation. (A) ELISA detection of LPS on LPS-coated plates. (B) AP complement activation by plate-bound S typhosa LPS in wild-type (WT) or properdin knockout (P−/−) mouse serum. To reconstitute AP activity in properdin−/− mouse serum, C3−/− serum or purified human properdin (hP) was premixed with properdin−/− mouse serum. Alternatively, LPS-coated plates were incubated with hP and washed (hP coat) before exposure to properdin−/− serum. Similar assays were performed with plate-bound LPS from S minnesota (S) (C) or E coli (D). (E,F) ELISAs of hP interaction with plate-bound LPS. Plates were first coated with different concentrations of LPS and then incubated with a fixed concentration of purified hP (62.5 ng/well; E). (F) Plates were first coated with a fixed concentration of LPS (5 μg/mL) and then incubated with increasing concentrations of purified hP. After washing, the amount of plate-bound properdin was detected by antiproperdin antibodies. OD, optical density.
By premixing with C3−/− mouse serum, we observed similar reconstitution of S minnesota (S) and E coli LPS-induced AP complement activity in properdin−/− serum (Figure 3C,D). Surprisingly, unlike the observation with S typhosa LPS, purified human properdin only partially restored S minnesota (S) and E coli LPS-induced AP complement activity in properdin−/− serum, irrespective of whether the protein was added to properdin−/− serum or used to pretreat LPS-coated plates (Figure 3C,D). We next compared the relative binding affinity of purified human properdin to LPS of the 3 bacteria species. Figure 3E,F shows that human properdin did not bind to the plate in the absence of LPS coating, but it displayed a clear LPS concentration– and properdin concentration–dependent binding to S typhosa LPS. This contrasted starkly with its weak binding to S minnesota (S) and E coli LPS. Thus, the ability of human properdin to restore LPS-dependent AP complement activity in properdin−/− mouse serum correlated with its binding affinity to LPS. Presumably, mouse properdin had equally high affinity toward S typhosa, S minnesota (S), and E coli LPS as C3−/− mouse serum was fully active at restoring S minnesota (S) and E coli LPS-induced, as well as S typhosa LPS-induced, AP complement activity to properdin−/− mouse serum (Figure 3B-D).
AP activation on nonprotected autologous cells is also dependent on properdin
We previously demonstrated that mouse erythrocytes deficient in Crry, a membrane-anchored complement inhibitory protein, were susceptible to AP complement–mediated extravascular hemolysis (phagocytosis) in vivo.19,20 To assess the role of properdin in this process, we transfused Crry-deficient mouse erythrocytes into WT and properdin−/− mice. Figure 4A shows that Crry-deficient erythrocytes were rapidly eliminated in WT but not properdin−/− recipients. Thus, spontaneous AP complement activation on nonprotected autologous cells also required properdin to be initiated.

Crry−/− erythrocytes– and zymosaninduced AP complement activation. (A) Survival of biotin–labeled Crry−/− mouse erythrocytes (109) in wild-type (WT) or properdin−/− mice. The percentage of Crry−/− erythrocytes in the recipient mouse 5 minutes after transfusion was determined by FACS and taken as 100%. (B) Representative FACS analysis of C3 deposition on zymosan after incubation with WT, properdin−/−, or factor B knockout (fB−/−) mouse serum in Mg2+-EGTA. (C) Quantitation of C3 deposition on zymosan. Experiments were performed with 2 serum dilutions (1:10, 1:20) and 2 zymosan concentrations (0.025 mg/mL, 0.125 mg/mL). N = 3 mice per group. Error bars represent standard deviations. MFI indicates mean fluorescence intensity. P values refer to Student t test.
Crry−/− erythrocytes– and zymosaninduced AP complement activation. (A) Survival of biotin–labeled Crry−/− mouse erythrocytes (109) in wild-type (WT) or properdin−/− mice. The percentage of Crry−/− erythrocytes in the recipient mouse 5 minutes after transfusion was determined by FACS and taken as 100%. (B) Representative FACS analysis of C3 deposition on zymosan after incubation with WT, properdin−/−, or factor B knockout (fB−/−) mouse serum in Mg2+-EGTA. (C) Quantitation of C3 deposition on zymosan. Experiments were performed with 2 serum dilutions (1:10, 1:20) and 2 zymosan concentrations (0.025 mg/mL, 0.125 mg/mL). N = 3 mice per group. Error bars represent standard deviations. MFI indicates mean fluorescence intensity. P values refer to Student t test.
Properdin is not essential for zymosan– or CVF–induced AP complement activation
The yeast cell wall component zymosan is another well-known AP complement activator.7 We incubated zymosan with WT or properdin−/− mouse serum and assessed AP complement activation by FACS analysis of C3 deposition. As shown in Figure 4B,C, we found that zymosan–induced AP complement activation was only partially impaired in properdin−/− serum. This was in clear contrast with factor B–deficient (fB−/−) mouse serum, which supported no AP complement activation (Figure 4B).
CVF binds factor B with high affinity and CVFBb acts as a stable C3 convertase to cause extensive AP complement activation in vivo and in vitro.24 To evaluate the role of properdin in CVF-induced AP complement activation, we treated WT or properdin−/− mouse serum with CVF and analyzed C3 activation kinetics by Western blot analysis. We found that CVF (0.01μg for 5 μL serum) induced complete C3 cleavage within 20 minutes in both types of sera, but the C3 activation kinetics in the properdin−/− serum appeared to be slightly delayed (Figure 5A-C). However, we observed no difference between WT and properdin−/− sera when a higher dose of CVF (0.3 μg for 5 μL serum) was used. In this case, complete C3 cleavage was achieved within 1 minute of CVF treatment in both sera (data not shown). Thus, properdin plays an insignificant role in CVF-induced AP complement activation.

CVF–induced AP and anti-OVA/OVA–induced classic pathway complement activation. Western blot analysis of C3 activation in wild-type (A) or properdin−/− (B) mouse serum. Cleavage product of the C3 α-chain was detected in serum treated with CVF in Mg2+-EGTA but not in untreated serum or serum treated with CVF in EDTA. (C) Densitometry of cleaved and intact C3 α-chain in panels A and B. (D) ELISA plate assays of anti-OVA/OVA–induced classic pathway complement activation in wild-type (WT), properdin−/−, and factor B knockout (fB−/−) mouse serum or in properdin−/− serum treated with an antihuman fB antibody.
CVF–induced AP and anti-OVA/OVA–induced classic pathway complement activation. Western blot analysis of C3 activation in wild-type (A) or properdin−/− (B) mouse serum. Cleavage product of the C3 α-chain was detected in serum treated with CVF in Mg2+-EGTA but not in untreated serum or serum treated with CVF in EDTA. (C) Densitometry of cleaved and intact C3 α-chain in panels A and B. (D) ELISA plate assays of anti-OVA/OVA–induced classic pathway complement activation in wild-type (WT), properdin−/−, and factor B knockout (fB−/−) mouse serum or in properdin−/− serum treated with an antihuman fB antibody.
Properdin plays a negligible role in classical pathway–triggered AP complement amplification
Activation of the classical and lectin pathways inevitably initiates the AP pathway. To determine whether properdin plays a role in classical pathway-triggered AP complement amplification, we used a plate-based assay to measure anti-OVA/OVA–induced complement activity in WT and properdin−/− sera.23 We used fB−/− serum as a negative control for AP amplification in this experiment. As shown in Figure 5D, we observed a significant difference in complement activation between WT and fB−/− mouse serum, confirming that AP amplification contributes substantially to the overall complement activation initiated via the classical pathway.31,32 In contrast, anti-OVA/OVA–induced complement activation was minimally reduced in properdin−/− serum (Figure 5D). This result suggested that either the AP amplification loop was largely intact in properdin−/− mice or there was compensatory up-regulation in the activity of the classical pathway C3 convertase. To distinguish these 2 possibilities, we depleted fB from properdin−/− serum using antihuman fB antibodies. We found that depletion of fB from properdin−/− serum reduced anti-OVA/OVA–induced complement activation to a level that was comparable to that observed in fB−/− serum (Figure 5D). This result established that the AP amplification loop was largely intact in properdin−/− mice.
Properdin and AP play a more significant role in LOS- than in LPS-induced complement activation in vivo
Human properdin–deficient patients are susceptible to lethal meningococcal infection.17 Because N meningitides bacteria contain LOS rather than LPS in their outer membranes, we examined the role of properdin in N meningitides LOS-induced complement activation in vitro and in vivo. Using LOS-coated plate assays in Mg2+-EGTA, we found that LOS, like LPS, induced AP complement activation in WT but not properdin−/− mouse serum (data not shown). Furthermore, by measuring plasma levels of activated C3, we found that injection of LOS caused systemic complement activation in WT but not properdin−/− mice in vivo (Figure 6A). Notably, we observed that LPS-induced systemic complement activation in vivo was reduced but not abolished in properdin−/− mice (Figure 6B). These results suggested that LOS activated complement in vivo principally via the AP pathway, whereas LPS activated complement through both AP-dependent and -independent pathways. Indeed, by performing LPS- or LOS-coated plate assays in GVB2+ buffer to enable all 3 complement activation pathways, we demonstrated that fB or properdin deficiency caused a much more dramatic reduction in LOS-induced complement activation than in LPS-induced complement activation (Figure 6C,D).

LOS- and LPS-induced complement activation in vivo and in vitro. ELISAs of plasma C3 activation products in wild-type (WT) and properdin−/− mice 1 hour after LOS (A) or LPS (B) treatment. LOS or LPS was given at 20 mg/kg (intraperitoneally) and phosphate-buffered saline was used as a vehicle control. N = 3 mice per group. Error bars represent standard deviations. A wild-type mouse plasma sample treated with CVF in vitro was used as a reference for C3 activation (100%). ELISA of LOS– (C) or LPS–induced (D) total complement activation in wild-type (WT), properdin−/−, or factor B knockout (fB−/−) mouse serum in GVB2+ buffer.
LOS- and LPS-induced complement activation in vivo and in vitro. ELISAs of plasma C3 activation products in wild-type (WT) and properdin−/− mice 1 hour after LOS (A) or LPS (B) treatment. LOS or LPS was given at 20 mg/kg (intraperitoneally) and phosphate-buffered saline was used as a vehicle control. N = 3 mice per group. Error bars represent standard deviations. A wild-type mouse plasma sample treated with CVF in vitro was used as a reference for C3 activation (100%). ELISA of LOS– (C) or LPS–induced (D) total complement activation in wild-type (WT), properdin−/−, or factor B knockout (fB−/−) mouse serum in GVB2+ buffer.
Discussion
In this study, we have created a properdin-deficient mouse model to characterize the role of properdin in complement activation. Although properdin was originally discovered and believed to be an initiator of the AP complement, the current view of properdin function is that it facilitates AP complement activation by stabilizing the nascent AP C3 convertase, C3bBb.10 However, to what degree AP complement activation depends on properdin and whether there is differential requirement of properdin by various AP complement activating surfaces has not been well defined. Related to these questions, it is not clearly understood why properdin-deficient patients are selectively predisposed to lethal meningococcal infection.17
We show here that properdin is indispensable for LPS- and LOS-induced AP complement activation and for AP complement-mediated extravascular hemolysis of Crry-deficient erythrocytes. On the other hand, we found that zymosan-induced AP complement activation was only moderately impaired by properdin deficiency. Furthermore, properdin appeared to play negligible, if any, role in CVF- and classical pathway–triggered AP complement amplification. These results support the following conclusions: (1) properdin is more relevant to independent AP complement initiation than AP complement amplification secondary to other activation pathways; (2) the need for properdin in AP complement initiation is variable and depends on the nature of the activating surface; and (3) both foreign and endogenous AP complement activators may critically depend on properdin for their activity.
Our findings presented here may help explain the fact that the traditional hemolytic assays for the alternative pathway did not usually detect human properdin deficiency, whereas the new LPS-based ELISAs (eg, Wielisa kit, http://www.wieslab.se) do detect this deficiency (Dr Tom Mollnes, University of Oslo, Norway, oral communication, June 2007). Two potential explanations could account for the differential requirement of properdin by AP complement activators. First, AP activation on a given surface represents the balance between properdin-dependent promotion via C3bBb stabilization and factor H (fH)–dependent inhibition of C3 “tick-over.” In this scenario, an AP activator for which properdin is not essential may have limited interaction with fH and, as a result of lacking sufficient fH-dependent inhibition, spontaneous C3 activation and amplification could occur as a default process without the help of properdin. Although this theory may explain the differential requirement of properdin in LPS- and zymosan-induced AP activation (ie, because of their differential interaction with fH), it could not explain why purified human properdin, unlike C3−/− serum as a source of mouse properdin, restored S. typhosa LPS-induced, but not S minnesota (S) or E coli LPS-induced, AP complement activity in properdin−/− serum (Figure 3). Clearly, nonspecific stabilization of C3bBb cannot completely account for the mechanism of action of properdin.
A second but not mutually exclusive theory is that properdin binds to an AP activator, either directly or via initially deposited C3b, and directs complement activation by serving as a platform for new C3bBb assembly. Indeed, surface-bound properdin was demonstrated to promote C3bBb formation in a recent biochemical study,11 and our results here showed that the ability of human properdin to restore LPS-induced AP complement activity in properdin−/− mouse serum correlated with its LPS-binding affinity. Furthermore, we demonstrated that LPS-bound human properdin was capable of activating AP complement in properdin−/− mouse serum in the absence of any solution properdin. Whether direct binding of properdin to an activator occurs and constitutes an obligatory step in AP complement initiation under physiologic conditions remains to be established. We attempted but could not demonstrate properdin binding to plate-bound S typhosa LPS using C3-depleted human serum, nor could we detect AP complement activity in properdin−/− mouse serum after pretreating S typhosa LPS-coated plates with C3−/− mouse serum as a source of properdin (data not shown). It is possible that the avidity of properdin binding to an AP activator such as LPS in whole serum was competitively inhibited by other serum components but was sufficient to induce initial C3b deposition, which in turn leads to more properdin binding via C3b.11
If, by virtue of its binding affinity toward an activating surface, properdin acts as an obligatory pattern recognition molecule for AP complement initiation, then the observation that zymosan caused vigorous AP complement activation in properdin−/− mouse serum raised the possibility of other, as yet to be identified, factor(s) that may act in a similar activator-specific manner for AP complement initiation. In this context, it is of interest to note an earlier study that putatively identified such an “initiation factor” in zymosan-induced AP complement activation that was distinct from properdin.33
That properdin plays an essential role in LPS- and LOS-induced AP complement activation is consistent with the known susceptibility of properdin-deficient patients to bacterial infection, but it does not explain their selective sensitivity to meningitides.17 Of note, we observed that LOS-induced complement activation in vivo was essentially abolished in properdin−/− mice, whereas that induced by LPS was only partially impaired. This suggested that AP was the predominant pathway in LOS- but not LPS-induced complement activation, a conclusion that was verified by in vitro assays of total complement activation in GVB2+ buffer. Thus, properdin deficiency, especially when combined with low antibody or mannose-binding lectin levels,14,15 may essentially abrogate complement–mediated bactericidal activity toward LOS-containing meningitides but have less significant consequences for the control of LPS-containing bacteria and other pathogens.
In conclusion, we have created and studied a properdin−/− mouse to arrive at several important conclusions that shed new light on the mechanism of action of properdin in AP complement activation. Apart from playing a role in host defense, properdin produced by leukocytes at sites of inflammation has been postulated to initiate AP complement and amplify tissue injury.4,34 The availability of the properdin−/− mouse described here will now also allow the direct testing of this hypothesis.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
We thank Dr Sanjay Ram for N meningitidis LOS, Dr Hector Molina for Crry−/−/C3−/− mice, and Dr John Lambris for anti-OVA, C3c antibodies, and useful discussions.
This work was supported by National Institutes of Health grants AI-62388, AI-49344, and AI-44970 and grant RG 3671-A-1 from the National Multiple Sclerosis Society.
National Institutes of Health
Authorship
Contribution: Y.K., T.M., L.Z., and W.-C.S. designed experiments and analyzed data; Y.K., L.Z., and T.M. performed experiments; and Y.K. and W.-C.S. wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Wen-Chao Song, Institute for Translational Medicine and Therapeutics and Department of Pharmacology, University of Pennsylvania School of Medicine, Rm 1254 BRBII/III, 421 Curie Blvd, Philadelphia, PA 19104; e-mail: song@spirit.gcrc.upenn.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal