

Abstract
Juvenile myelomonocytic leukemia (JMML) is a rare pediatric myeloid neoplasm characterized by excessive proliferation of myelomonocytic cells. When we investigated the presence of recurrent molecular lesions in a cohort of 49 children with JMML, neurofibromatosis phenotype (and thereby NF1 mutation) was present in 2 patients (4%), whereas previously described PTPN11, NRAS, and KRAS mutations were found in 53%, 4%, and 2% of cases, respectively. Consequently, a significant proportion of JMML patients without identifiable pathogenesis prompted our search for other molecular defects. When we applied single nucleotide polymorphism arrays to JMML patients, somatic uniparental disomy 11q was detected in 4 of 49 patients; all of these cases harbored RING finger domain c-Cbl mutations. In total, c-Cbl mutations were detected in 5 (10%) of 49 patients. No mutations were identified in Cbl-b and TET2. c-Cbl and RAS pathway mutations were mutually exclusive. Comparison of clinical phenotypes showed earlier presentation and lower hemoglobin F levels in patients with c-Cbl mutations. Our results indicate that mutations in c-Cbl may represent key molecular lesions in JMML patients without RAS/PTPN11 lesions, suggesting analogous pathogenesis to those observed in chronic myelomonocytic leukemia (CMML) patients.
Introduction
Juvenile myelomonocytic leukemia (JMML) is a special subtype of myelodysplastic syndrome/myeloproliferative disorder (MDS/MPD) that, analogous to chronic myelomonocytic leukemia (CMML), is characterized by excessive proliferation of myelomonocytic cells, but unlike CMML it occurs in young children and shows characteristic hypersensitivity to granulocyte-macrophage colony-stimulating factor (GM-CSF).1-3 Mutations of genes involved in GM-CSF signal transduction, including RAS and PTPN11, can be identified in a majority of children with JMML.3-5 Constitutional mutations of NF1 can be found in another 10% of patients with JMML.1,6,7 Recent studies show that a common mechanism of NF1 inactivation is uniparental disomy (UPD) resulting in duplication of the mutant NF1 allele.7,8 NF1 is a GTPase activating protein for RAS and thereby acts as a tumor suppressor.9 Oncogenic RAS mutations at codons 12, 13, and 61 have been identified in approximately 20% to 25% of patients with JMML.4,10 These mutations lead to elevated levels of RAS-GTP, the active form of RAS.11 Somatic mutations in PTPN11, coding for tyrosine phosphatase Src homology 2 domain-containing protein, have been reported in 35% of patients with JMML,5,12,13 and induce hematopoietic progenitor hypersensitivity to GM-CSF due to hyperactivation of the RAS signaling axis.14,15
Based on the proposed paradigm that recurrent areas of somatic copy-neutral loss of heterozygosity can point toward the presence of homozygous mutations contained within the corresponding region,16 we have identified various recurrent areas of acquired segmental UPD, in particular in patients with MDS/MPD, including CMML. Such analyses have shown that, in addition to the recently identified Jak2V617F mutation associated with UPD9p, other known mutations can be duplicated by homologous recombination, including, for example, c-Mpl (UPD1p), FLT-3 ITD (UPD13q), TET2 (UPD4q), and others.17-22 Based on the observation of recurrent somatic UPD11q23.3, we have discovered homozygous c-Cbl mutations in the RING finger domain (RFD) occurring frequently in MDS/MPD and especially CMML or secondary acute myeloid leukemia (AML) evolved from CMML.23 When we analyzed other members of Cbl gene family, mutations were also found in Cbl-b and Cbl-c and were associated with an indistinguishable clinical phenotype.24 The Cbl gene family codes for E3 ubiquitin ligases (ULs) with the ubiquitination activity mediated via the RFD. They are involved in degradation of activated phosphotyrosine receptors and other phosphotyrosine kinases such as ζ-chain–associated protein kinase 70 involved in signal transduction.25 Thus, mutations in the RFD can lead to decreased receptor degradation and, analogous to PTPN11 mutations, result in augmentation of proliferative signals mediated by various growth factor receptors. In a c-Cbl−/− mouse model a mild myeloproliferative phenotype with expansion of stem cells and hyperresponsiveness to growth factors is found,26 whereas a RFD mutant knock-in model shows a severe myeloproliferative phenotype (W. Langdon, University of Western Australia, oral communication, January 2009). These observations, together with the transforming effects of the v-Cbl oncogene lacking the RFD, suggest that E3 UL activity is essential for the tumor suppressor function of c-Cbl, whereas the N-terminal portion of the protein may be oncogenic.
Based on the morphologic similarities of JMML and typical CMML, presence of growth factor hypersensitivity, and observation of UPD11q in children affected by JMML, we hypothesized that Cbl family mutations may also be present in a subset of patients with JMML. Here, we investigated 49 JMML patients with the goals of (1) identifying pathogenic molecular lesions, including mutations in Cbl gene family members, and (2) correlating clinical outcomes to presence and location of other pathogenic molecular lesions, including PTPN11, NRAS, KRAS, and TET2. Of note is that during review of our paper, c-Cbl mutations were reported in JMML.27
Methods
Patients
We studied 49 children (32 boys and 16 girls; 1 patient's sex was unknown) with JMML diagnosed between 1988 and 2008 in 28 institutions throughout Japan. Written informed consent for sample collection was obtained at appropriate institutions from patients' parents according to the institutional protocols and the Declaration of Helsinki. The sample repository was located at Nagoya University Graduate School of Medicine. Molecular analysis of the mutational status was approved by the Ethics Committee of Nagoya University Graduate School of Medicine. The diagnosis of JMML was based on the internationally accepted criteria previously published.26 We excluded patients with Noonan syndrome. The clinical and hematologic characteristics of the patients are summarized in Table 1. The median age at diagnosis was 28 months (range, 1-75 months). Karyotypic abnormalities were detected in 11 patients, including 7 patients with monosomy 7. Two children had clinical evidence of NF1. Of 49 patients, 32 underwent hematopoietic stem cell transplantation.
Characteristics of JMML patient cohort
| Variable | Total cohort, N = 49 |
|---|---|
| Median age at diagnosis, mo (range) | 32 (1-75) |
| Sex, male/female/unknown | 32/16/1 |
| NF1 by clinical diagnosis, yes/no | 2/47 |
| Median Hb, g/L (range) | 0.96 (0.49-1.20) |
| Median HbF, % (range) | 23.6 (1.0-62.0) |
| Median WBC, ×109/L (range) | 28.0 (10.9-126.2) |
| Median monocyte in PB count, ×109/L (range) | 4.5 (1.0-31.6) |
| Median plt, ×109/L (range) | 49 (1.4-320) |
| Metaphase cytogenetics, no. of patients (%) | |
| Normal karyotype | 35 (71.4) |
| Monosomy 7 | 8 (16.3) |
| Trisomy 8 | 1 (2.0) |
| Other abnormalities | 3 (6.1) |
| Unknown | 2 (4.1) |
| Hematopoietic stem cell transplantation | |
| Yes | 32 |
| No | 13 |
| Unknown | 4 |
| Status at last follow-up | |
| Alive | 24 |
| Dead | 21 |
| Unknown | 4 |
| Median observation period, mo (range) | 14 (1-216) |
| Variable | Total cohort, N = 49 |
|---|---|
| Median age at diagnosis, mo (range) | 32 (1-75) |
| Sex, male/female/unknown | 32/16/1 |
| NF1 by clinical diagnosis, yes/no | 2/47 |
| Median Hb, g/L (range) | 0.96 (0.49-1.20) |
| Median HbF, % (range) | 23.6 (1.0-62.0) |
| Median WBC, ×109/L (range) | 28.0 (10.9-126.2) |
| Median monocyte in PB count, ×109/L (range) | 4.5 (1.0-31.6) |
| Median plt, ×109/L (range) | 49 (1.4-320) |
| Metaphase cytogenetics, no. of patients (%) | |
| Normal karyotype | 35 (71.4) |
| Monosomy 7 | 8 (16.3) |
| Trisomy 8 | 1 (2.0) |
| Other abnormalities | 3 (6.1) |
| Unknown | 2 (4.1) |
| Hematopoietic stem cell transplantation | |
| Yes | 32 |
| No | 13 |
| Unknown | 4 |
| Status at last follow-up | |
| Alive | 24 |
| Dead | 21 |
| Unknown | 4 |
| Median observation period, mo (range) | 14 (1-216) |
NF1 indicates neurofibromatosis type1; Hb, hemoglobin; WBC, white blood cell; PB, peripheral blood; and plt, platelet.
SNP-A karyotyping analysis
Mononuclear cells were isolated using Ficoll-Hypaque density gradient centrifugation and cryopreserved until use. Genomic DNA was extracted using the QIAamp DNA Blood Mini Kit (QIAGEN). High-density Affymetrix single nucleotide polymorphism array (SNP-A; 250 K) was applied as a karyotyping platform to identify loss of heterozygosity (LOH), microamplification, and microdeletion as previously described.28
Bioinformatic analysis
Signal intensity was analyzed and SNP calls were determined using Gene Chip Genotyping Analysis Software Version 4.0 (GTYPE). Copy number and areas of UPD were investigated using a Hidden Markov Model and CN Analyzer for Affymetrix GeneChip Mapping 250-K arrays (CNAG Version 3.0) as previously described.28
We excluded germline-encoded copy number variation and nonclonal areas of gene copy number–neutral LOH from further analysis using a bioanalytic algorithm based on lesions identified by SNP-A in an internal control series (N = 713) and reported in the Database of Genomic Variants (http://projects.tcag.ca/variation).29 Through calculation of their average sizes, we defined a maximal size of germline LOH in controls and consequently excluded all defects of this type in patients' samples; according to 95% confidence interval, stretches of UPD larger than 25.8 Mb were considered unlikely of germline origin. In addition, all nonclonal areas of UPD seen in controls were interstitial.
PTPN11, NRAS, KRAS, TET2, and E3 ubiquitin ligase mutational screening
To screen for PTPN11 mutations, we polymerase chain reaction amplified genomic DNA corresponding to exons 2, 3, 4, 7, 8, 12, and 13 as previously reported.12,30,31 NRAS and KRAS mutations in codons 12, 13, and 61 were identified as previously described32,33 and were confirmed by sequencing. To screen patients for mutations in E3 ubiquitin ligase genes and TET2, direct genomic sequencing of exons constituting the RFD of Cbl family members (exons 8 and 9 of c-Cbl, exons 9 and 10 of Cbl-b, exons 7 and 8 of Cbl-c, and exons 3-11 of TET2) was performed. For sequencing, 250 ng of polymerase chain reaction product, 3μM original forward or reverse primer, 2 μL of Big Dye Version 3.1 (Applied Biosystems), and 14.5 μL of deionized H2O were amplified under the following conditions: 95°C (2 minutes) followed by 25 cycles of 95°C (10 seconds), 50°C (5 seconds), and 60°C (4 minutes). Sequencing was performed as previously described.22
GM-CSF hypersensitivity assay
GM-CSF hypersensitivity assays were established as described previously.2 Briefly, we used cytokine-free methocult H4230 (StemCell Technologies), and added 1 × 103 CD34+ bone marrow cells that were prepared by positive selection with magnetic-activated cell sorting beads (Miltenyi Biotec). Recombinant human GM-CSF (R&D Systems) was added at the time the cultures were initiated. Cultures were performed in duplicate, and colonies of 40 or more cells were scored after 14 days of incubation. The data are expressed as percentage of maximal numbers of granulocyte-macrophage colony-forming units (CFU-GMs). This approach more accurately reflects changes in sensitivity and does not bias the results compared with graphing actual counts because most JMML samples had considerably higher total numbers of CFU-GMs than controls, although there was considerable patient-to-patient variability.
Statistical analysis
When appropriate, Kaplan-Meier statistics were applied to assess survival. For comparison of the frequency of mutation or other clinical features between disease groups, categoric variables were analyzed using the Fisher exact test and continuous variables were tested using the Mann-Whitney U test.
Results
Cytogenetic and clinical characterization of JMML patients
First, we performed SNP-A– and metaphase cytogenetics–based analyses. Using conventional metaphase cytogenetics, chromosomal aberrations were found only in a minority of patients (25%). SNP-A–based karyotyping confirmed the results of metaphase cytogenetics, including the presence of monosomy 7 in 7 patients and trisomy 8 in 1 patient. However, due to increased precision and ability to detect copy-neutral loss of heterozygosity of SNP-A, additional lesions were identified by SNP-A in 24 (49%) of 48 patients, including trisomy 21 not detected by metaphase cytogenetics (MC) in 1 patient and microdeletions in 9 patients (Figure 1A), including 1q25.3 (patient 44), 2p22.1 (patient 46), 5q23.1 (patient 20), 5q31.3 (patient 40), 6q21q25.3 (patient 15), 8p21.2 (patient 36), 12p13.2 (patient 3), 17q11.2 (patients 15, 49), and 19p13.3 (patient 2). We also detected microamplifications in 7 patients (Figure 1A), located at 1p31.1 (patient 14), 1q44 (patient 39), 7p21.1 (patient 16), 7q11.22 (patient 17), 10p11.23 (patient 29), 15q26.3 (patient 47), and 18q12.3 (patient 22). The shared copy number–altering lesions included monosomy 7 and loss of 17q11.2, which contained the NF1 locus. Although we were unable to confirm the somatic nature of the submicroscopic defects due to lack of germline DNA, these lesions did not overlap with copy number variations present in internal control cohort and publicly available databases. Most significantly, we identified UPD in 5 patients (Figure 1A). UPD11q was found in 4 patients, all regions overlapping from 11q23.3 to the telomere. This commonly affected region contained the c-Cbl locus (Figure 1A,C). The region of UPD at 17q contained the NF1 locus and corresponded with clinical neurofibromatosis features. Overall, compared with the results of MC, SNP-A identified significantly more genetic abnormalities (25% vs 49%; P = .02; Figure 1B).

Single nucleotide polymorphism array–based karyotyping of JMML. (A) Genomic distribution and type of lesion identified in patients with JMML by SNP-A analysis. Green bar represent amplification, red shows deletion, and blue corresponds to UPD. Red lines pinpoint the locus of genes discussed in the text, as well as the number of patients mutated at that locus. NF1 mutational status was not assessed in this cohort. (B) Increased sensitivity of SNP-A for detecting chromosomal lesions. The results of MC (25%) and by SNP-A (49%) from the JMML cohort studied are shown. (C) Representative 250-K SNP-A analysis of UPD11q by CNAG Version 3.0 (patient 16). Both the raw and averaged total copy number (CN) tracks (red dots, blue line) show a normal copy number, whereas heterozygous SNP calls and allele-specific copy number tracks (green dashes, red/green lines) show a reduction in copy number, indicating UPD. The specific localization of 11qUPD in 4 patients (patients 16, 27, 38, and 43) is indicated by the blue bars. The c-Cbl locus is indicated on the chromosome 11 idiogram with a yellow line.
Single nucleotide polymorphism array–based karyotyping of JMML. (A) Genomic distribution and type of lesion identified in patients with JMML by SNP-A analysis. Green bar represent amplification, red shows deletion, and blue corresponds to UPD. Red lines pinpoint the locus of genes discussed in the text, as well as the number of patients mutated at that locus. NF1 mutational status was not assessed in this cohort. (B) Increased sensitivity of SNP-A for detecting chromosomal lesions. The results of MC (25%) and by SNP-A (49%) from the JMML cohort studied are shown. (C) Representative 250-K SNP-A analysis of UPD11q by CNAG Version 3.0 (patient 16). Both the raw and averaged total copy number (CN) tracks (red dots, blue line) show a normal copy number, whereas heterozygous SNP calls and allele-specific copy number tracks (green dashes, red/green lines) show a reduction in copy number, indicating UPD. The specific localization of 11qUPD in 4 patients (patients 16, 27, 38, and 43) is indicated by the blue bars. The c-Cbl locus is indicated on the chromosome 11 idiogram with a yellow line.
Mutational analysis of patients with JMML
After defining chromosomal defects associated with JMML, we performed mutational analysis of the genes known to be affected by mutations in JMML. PTPN11 mutations were found in 26 (53%) of 49, whereas NRAS and KRAS mutations were found in 2 (4%) of 49 and 1 (2%) of 49, respectively (Table 2). None of the patients screened show the presence of TET2 mutations, previously shown to be present in a significant proportion of patients with MDS/MPD, including CMML.21 Excluding patients with a neurofibromatosis phenotype, 18 (37%) of 49 of patients did not show any of the known pathogenic defects occurring in JMML.

Identification of Cbl gene family mutations in JMML
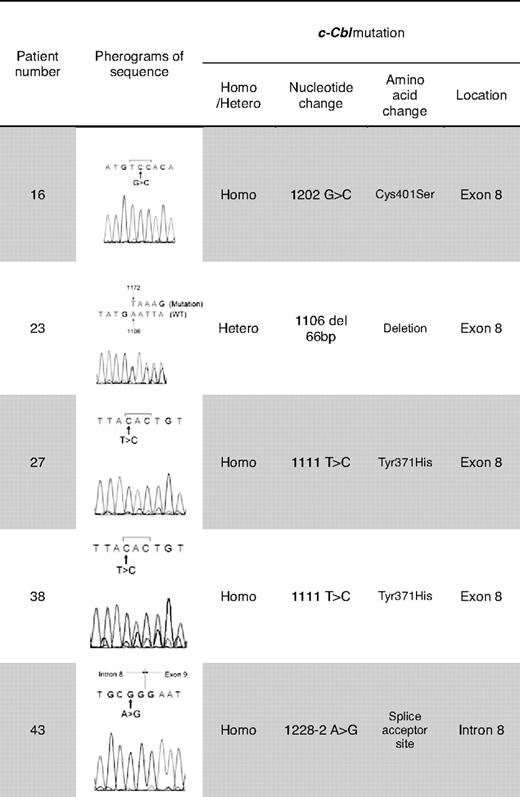
Previously, homozygous c-Cbl mutations in the RFD were identified in patients with MDS/MPD, especially CMML or secondary AML that evolved from CMML.22 We focused our attention on this gene as UPD11q was found in 4 of 49 JMML patients. Mutational analysis of Cbl family genes revealed mutations of c-Cbl in 5 (10%) of 49 patients, and no Cbl-b mutations (Tables 2–3). c-Cbl mutations were heterozygous in 1 patient (patient 23) and homozygous in 4 patients (patients 16, 27, 38, 43; supplemental Table 1, available on the Blood website; see the Supplemental Materials link at the top of the online article). All mutations were located in the RFD (exon 8 and intron 8); 2 patients had an identical homozygous mutation (1111T>C, Tyr371His; patients 27, 38). All 4 patients with a homozygous c-Cbl mutation simultaneously harbored UPD11q (supplemental Table 1, Figures 1C-2). In addition, no patient with a c-Cbl mutation had mutations in genes known to play a role in JMML (PTPN11, NRAS, and KRAS; Table 2) or had clinical diagnosis of NF1. Excluding patients with a neurofibromatosis phenotype, 13 (26.5%) of 49 of patients did not have the mutation of PTPN11, RAS, and c-Cbl genes.

Site of the c-Cbl mutations and predicted product of splice variant in the intron 8 splice acceptor site. (A) Localization of the c-Cbl mutations within the predicted protein product. Red arrows show the site of mutations in exon, and blue arrows show the site of splice variant. (B) In patient 48, a homozygous mutation was seen in the intron 8 splice acceptor site of c-Cbl. According to http://genome.cbs.dtu.dk/services/NetGene2,34 this mutation may result in a splice variant, leading to a shorter transcript in RF domain.
Site of the c-Cbl mutations and predicted product of splice variant in the intron 8 splice acceptor site. (A) Localization of the c-Cbl mutations within the predicted protein product. Red arrows show the site of mutations in exon, and blue arrows show the site of splice variant. (B) In patient 48, a homozygous mutation was seen in the intron 8 splice acceptor site of c-Cbl. According to http://genome.cbs.dtu.dk/services/NetGene2,34 this mutation may result in a splice variant, leading to a shorter transcript in RF domain.
When we investigated our cohort for the presence of Cbl-c mutations, we found heterozygous frameshift nucleotide variation (1256 insertion C; patients 7, 12, 14, 29, 33, 38, and 46; data not shown). However, Cbl-c mutational status of germline though sequencing of nonclonal CD3+ lymphocytes in those patients showed the same frameshift mutation. Consequently, these Cbl-c nucleotide exchanges represent rare polymorphisms.
GM-CSF hypersensitivity assay
We also investigated whether JMML-specific GM-CSF is related exclusively to individual types of mutations identified, including c-Cbl mutations. CD34+ bone marrow cells' colony counts are expressed as percentage of maximal (supraoptimal) number of CFU-GMs (colony counts at any given concentration of GM-CSF/colony counts at 10 ng/mL GM-CSF). The colony growth of JMML cells with or without c-Cbl mutation did not differ from normal controls in low concentration of GM-CSF. For example at 0.01 ng/mL GM-CSF, colony counts were 55% (± 8%) with c-Cbl mutation (n = 4) versus 65% (± 10%) without c-Cbl mutation (n = 14) versus 15% (± 5%) in controls (n = 2; P = .042). At 0.1 ng/mL GM-CSF, colony counts were 87% (± 6%) versus 83% (± 11%) versus 15% (± 5%; P = .011) and at 1.0 ng/mL GM-CSF, 94% (± 11%) versus 93% (± 7%) versus 43% (± 3%; P = .063), respectively. Consequently, our results indicate that GM-CSF hypersensitivity of CD34+ cells from JMML patients may be a result of various molecular lesions including c-Cbl mutations (Figure 3).

GM-CSF hypersensitivity assay. Colony counts are expressed as percentage of maximal numbers of CFU-GM (colony counts cultured with each concentration of GM-CSF/colony counts cultured with 10 ng/mL GM-CSF). The similar GM-CSF hypersensitivity was seen in JMML patients with or without c-Cbl mutation. Error bars represent SE.
GM-CSF hypersensitivity assay. Colony counts are expressed as percentage of maximal numbers of CFU-GM (colony counts cultured with each concentration of GM-CSF/colony counts cultured with 10 ng/mL GM-CSF). The similar GM-CSF hypersensitivity was seen in JMML patients with or without c-Cbl mutation. Error bars represent SE.
Clinical features associated with Cbl gene family mutations
Although different molecular lesions can result in similar clinical phenotypes, specific mutations can modify clinical behavior and morphologic features. Consequently, we analyzed clinical characteristics of patients with specific mutations (Table 4).
Comparison of clinical characteristics for JMML patients with and without c-Cbl mutation
| Variable | With c-Cbl mutation, n = 5 | Without c-Cbl mutation, n = 44 | P |
|---|---|---|---|
| Median age at diagnosis, mo (range) | 12 (8-15) | 29 (1-75) | .037 |
| Median HbF, % (range) | 3.5 (2.0-7.6) | 24.9 (1.0-62.0) | .02 |
| Variable | With c-Cbl mutation, n = 5 | Without c-Cbl mutation, n = 44 | P |
|---|---|---|---|
| Median age at diagnosis, mo (range) | 12 (8-15) | 29 (1-75) | .037 |
| Median HbF, % (range) | 3.5 (2.0-7.6) | 24.9 (1.0-62.0) | .02 |
Other variables studied (sex, hemoglobin level, white blood cell count, platelet count, monocyte percentage in peripheral blood, and metaphase cytogenetic abnormalities) do not show statistical significance.
HbF indicates hemoglobin F.
We did not find any distinctive morphologic features of patients with Cbl gene family mutations and no differences were present in the blood counts at initial presentation. Other variables studied (sex, the presence of cytogenetic abnormalities) also did not differ between patients grouped according to mutational status. However, patients with mutant c-Cbl compared with those with wild-type constellation showed earlier presentation (median age at diagnosis, 12 months vs 29 months, P = .037) and lower median hemoglobin F (HbF) percentage (3.5% vs 24.9%, P = .02), previously shown to correlate with less favorable prognosis.1,33,35-40 Low HbF values in c-Cbl mutant cases were not attributable to monosomy 7, absent in this patient cohort. The probability of 2-year overall survival of c-Cbl mutant patients (50.0%; 95% confidence interval [CI], 25.0%-75.0%; n = 4) was similar to that of patients without c-Cbl mutations (50.4% [95% CI, 42%-59%]; n = 41). Similarly, when patients with all Cbl gene family mutations were analyzed, no distinct clinical features including differences in outcomes were found.
Discussion
The molecular pathogenesis of the often heterogeneous myeloid malignancies is not discernable through traditional morphologic analyses. Conversely, various molecular mechanisms can lead to similar clinical phenotypes and distinct mutational steps can result in various types of functional defects, each requiring distinct therapeutic approaches. Although JMML is associated with mutations in PTPN11 and RAS in a large proportion of cases3-5,12,13 and mutations of NF-1 in a smaller fraction,1,6,7 no specific mutations can be identified in a number of children affected by this disease.
Previously, we identified UPD11q and associated homozygous c-Cbl mutations in patients with CMML and secondary AML with monocytoid features.23 We have also noted that heterozygous mutations of other closely related E3 ULs such as Cbl-b and Cbl-c may be found in some patients with otherwise indistinguishable morphologic features; these mutations presented in heterozygous constellation as they were not associated with corresponding areas of somatic UPD.24 We have also found a significant proportion of CMML cases with UPD4q and microdeletions corresponding to the location of TET2 gene. We have shown that UPD4q is associated with TET2 mutations but, unlike for c-Cbl, heterozygous TET2 mutations were common.22
Based on our progress in CMML, in this article we undertook the molecular analysis of cytogenetic abnormalities and mutational events in the clinically similar syndrome of JMML occurring in children. Using SNP-A analysis we show that patients with JMML, in addition to known typical chromosomal defects, harbor invariant somatic copy-neutral loss of heterozygosity, in particular UPD11q23.3. Based on this finding and the previously shown association of UPD11q with c-Cbl mutation, we demonstrated that c-Cbl mutations located in the RFD of this gene are found in 5 (10%) of 49 of JMML patients. Since submission of this paper, similar results were reported by Loh et al.27 Unlike in adult CMML, TET2 mutations were not identified in JMML, a finding consistent with the absence of UPD4q or del4 in JMML.
Our findings suggest that selective pressure in JMML leads to use of functionally related pathways but may involve distinct genes. In fact, both c-Cbl (ubiquitination) and PTPN11 (dephosphorylation) mutations can lead to the augmentation of growth factor receptor–mediated signals and may explain why GM-CSF hypersensitivity is present in patients with JMML irrespective of whether c-Cbl, PTPN11, or RAS is mutated.
For Cbl mutations, in addition to the impaired degradation of activated growth factor receptors, altered ζ-chain–associated protein kinase 70 activation by c-Cbl may mediate proliferative signals analogous to RAS. Moreover, by binding to Grb2, c-Cbl competes with the guanine-nucleotide-exchange factor son-of-sevenless, thereby blocking signaling through the RAS–mitogen-activated protein kinase pathway and inhibiting proliferation.25 In agreement with this theory, RFD mutant knock-in mouse experiments suggest that c-Cbl deprived of its E3 ligase activity may act as an oncogene, and functional analysis of mutated c-Cbl showed that mutated c-Cbl has an oncogenic effect.35 These findings conclusively prove the pathogenic role of c-Cbl mutation in hematologic malignancies.
Our earlier studies showed that c-Cbl mutations stem from a somatic event and are not present in germline23 ; however, germline DNA was not available from our patients to conduct confirmatory studies. Nevertheless, c-Cbl mutations in JMML were similar or identical to those previously shown in CMML, for which the somatic nature has been confirmed through analysis of germline DNA and serial studies. Similarly, c-Cbl mutations were present exclusively in the context of UPD11q23.3, shown to occur only as a clonal somatic event. In agreement with a previous report,7 we have also found UPD17q in association with neurofibromatosis-associated JMML.
Patients with c-Cbl mutations show comparable survival as those without c-Cbl mutations, but a large fraction of these patients underwent transplantation. However, c-Cbl mutations were associated with a younger age of presentation and smaller percentage of HbF. Given that in previous reports an older age at diagnosis and elevated HbF level have been repeatedly described as risk factors for survival in JMML,1,36-42 lack of these poor prognostic markers in c-Cbl patients who demonstrate a similar outcome argues for an unfavorable impact of c-Cbl mutation, analogous to adult patients with c-Cbl.
In summary, our study describes a novel molecular lesion in children affected by JMML, suggesting similarity in the pathogenesis of a portion of patients with JMML to those with CMML.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported in part by the National Institutes of Health (R01 HL-082983, U54 RR019391, K24 HL-077522), and a grant from Aplastic Anemia & Myelodysplastic Syndromes International Foundation and Robert Duggan Charitable Fund (J.P.M).
National Institutes of Health
Authorship
Contribution: H. Muramatsu and H. Makishima designed research, performed research, analyzed data, and wrote the paper; A.M.J. and H.C. performed research; C.O. designed research, analyzed data, and wrote the paper; N.Y., Y.X., N.N., A.H., H.Y., Y.T., K.K., and A.M. designed research; S.K. designed research and wrote the paper; and J.P.M. designed research, performed research, analyzed data, and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Jaroslaw P. Maciejewski, Taussig Cancer Center/R40, 9500 Euclid Ave, Cleveland, OH 44195; e-mail: maciejj@ccf.org.
