

Abstract
Glucocorticoids are used extensively to treat autoimmune hemolytic anemias. Some beneficial effects of glucocorticoid pulse therapy have also been reported in sickle cell disease and paroxysmal nocturnal hemoglobinuria. Based on established concepts of hemoglobin (Hb) toxicity and physiologic Hb scavenger systems, we evaluated whether glucocorticoids could support an adaptive response to extracellular Hb independently of their immunosuppressive activities. Using global proteome and transcriptome analysis with mass-spectrometry (isobaric tag for relative and absolute quantitation and liquid chromatography-mass spectrometry) and gene-array experiments, we found that glucocorticoid treatment in vitro and in patients on glucocorticoid-pulse therapy polarized monocytes into a M2/alternatively activated phenotype with high Hb-scavenger receptor (CD163) expression and enhanced Hb-clearance and detoxification capability. Monocytes concurrently exposed to the interactive activity of glucocorticoids and extracellular Hb were characterized by high expression of a group of antioxidant enzymes known to be regulated by the conserved oxidative response transcription factor nuclear factor E2-related factor. Further, suppressed transferrin receptor, together with high ferroportin expression, pointed to a shift in iron homeostasis directed toward an increased cellular export of heme-derived iron. Therefore, stimulating Hb-endocytosis by CD163 and enhancing antioxidative homeostasis and iron recycling may be an essential activity of glucocorticoids that helps alleviate the adverse effects of extracellular Hb.
Introduction
Glucocorticoids are used extensively to treat inflammatory and autoimmune diseases. In autoimmune hemolytic anemia, glucocorticoids are thought to attenuate the production of erythrocyte-directed autoantibodies while reducing destruction of immunoglobulin-coated red blood cells by macrophages within the reticuloendothelial system.1 Intriguingly, randomized clinical trials demonstrated that glucocorticoid pulse therapy also has short-term beneficial effects on complications of nonimmune hemolysis such as acute chest syndrome and pain crisis in children with sickle cell disease.2,3 Likewise, glucocorticoid treatment of paroxysmal nocturnal hemoglobinuria (PNH) has been advocated to attenuate the severity of hemolytic crisis by not yet established mechanisms.4 Glucocorticoid treatment of PNH and sickle cell complications is controversial due to the high incidence of treatment-associated complications and rebound attacks.4-6 Nevertheless, these studies could suggest that glucocorticoids might enhance the level of protection against extracellular hemoglobin (Hb) exposure that occurs during nonimmune intravascular hemolysis in PNH and sickle cell disease. In addition, in autoimmune hemolytic anemia plasma, haptoglobin (Hp) is usually consumed pointing to the leakage of free Hb into the circulation.
Extracellular Hb has been associated with toxic adverse effects.7 Free radicals and altered Hb products are generated during the reaction between Hb-heme and physiologic oxidants such as H2O2.8 Depletion of vasodilatory nitric oxide is another extensively studied mechanism by which extracellular Hb can mediate endothelial dysfunction and vasoactivity.7 The latter mechanism probably plays a role in complications associated with occasional high plasma Hb concentrations observed during PNH crisis. However, there is significant controversy about the role of nitric oxide depletion in the chronic complications of sickle cell disease such as pulmonary hypertension.7,9 A third mechanism of Hb toxicity involves inflammatory and toxic processes that can be triggered by heme.10-12
Dedicated Hb scavenger and detoxification systems might represent specific targets for therapeutic intervention in hemolysis. Hp provides the first line of defense against extracellular Hb.13 When bound to Hp, Hb remains sequestered within the intravascular compartment, thus attenuating Hb-mediated vasoactivity, oxidative toxicity, and tissue peroxidation.14,15 Upon complex formation, Hp transforms the Hb αβ-dimer into a high-affinity ligand of the Hb scavenger receptor CD163, which then clears the Hb:Hp complex.16 CD163 is expressed exclusively by circulating blood monocytes and tissue macrophages. Because CD163+ circulating blood monocytes are continuously present at potential sites of intravascular hemolysis, these cells may play a unique role in the control of extracellular Hb toxicity.17
Earlier evidence suggests that glucocorticoids can positively regulate Hp and CD163.14,18-21 Therefore, this study systematically examined whether glucocorticoids also enhance cellular homeostatic mechanisms that finally could control Hb toxicity. Using a combined proteomics and transcriptomics approach, we found that glucocorticoid treatment in vitro and in vivo shifts monocyte differentiation toward a phenotype with a high Hb clearance and detoxification capacity. These changes were associated with stimulation of specific antioxidant and iron homeostasis pathways. Therefore, a shift of monocyte functions could represent a novel and essential activity of glucocorticoids, independent of and complementary to their well-understood anti-inflammatory and immunosuppressive characteristics.
Methods
Monocyte isolation and culture
Human-blood–derived macrophages were prepared from buffy coats of healthy donors that were purchased from the Swiss Red Cross Blood Bank (Swiss Red Cross; written informed consent was obtained from all donors in accordance with the Declaration of Helsinki), as described previously.22 Studies were approved by the University Hospital of Zurich ethics review board. The monocytes were cultured in Iscove modified Dulbecco medium (Invitrogen) supplemented with 10% heat-inactivated, pooled human serum that was prepared to have negligible residual free Hb. Alternatively, in some confirmatory experiments, medium was supplemented with 50% autologous serum as a source of endogenous Hp (approximately 1 mg/mL Hp). For glucocorticoid priming, monocytes were incubated for 36 hours with dexamethasone (Sigma-Aldrich) at a concentration of 2.5 × 10−7 M. Purified human Hp (mixed phenotype) was a kind gift of Benesis Corp. A highly purified and endotoxin-free human HbA0 was a kind gift of Hemosol.17 For confirmatory experiments performed in autologous serum, we used another endotoxin-free human Hb that was a kind gift of Robert Winslow (Sangart Inc). α-α DBBF crosslinked Hb (stabilized tetramer) was from Dr. Alayash (CBER, Food and Drug Administration).
iTRAQ labeling
At the end of experiments, cells were washed extensively in EDTA (ethylenediaminetetraacetic acid)–phosphate-buffered saline (PBS) to remove extracellular proteins and then lysed in CelLytic-M reagent (Sigma-Aldrich) supplemented with protease inhibitor (Roche Diagnostics). After 3 freeze-thaw cycles, cellular debris was removed by centrifugation at 16 000g for 10 minutes. Protein concentration of each sample was determined using a Bradford assay (Bio-Rad). Samples were normalized to 60 μg of protein, precipitated by addition of 1 volume of trichloroacetic acid to 4 volumes of protein to a final concentration of 10%-12% wt/vol trichloroacetic acid, and incubated at 4°C for 10 minutes. After centrifugation at 14 000 rpm, the supernatant was removed. The pellets were washed 3× with ice-cold acetone and dried in a heat block at 95°C for 10 minutes. Isobaric tag for relative and absolute quantitation (iTRAQ) labeling was performed according to the manufacturer's protocol (Applied Biosystems).
Strong cation exchange chromatography
iTRAQ-labeled peptides were fractionated by strong cation exchange liquid chromatography (SCX) using a polysulfoethyl A 2.1 × 200 mm, 5 μm, 300 Å column (PolyLC). Solvent A was 10mM potassium diphosphate, 25% acetonitrile (ACN), pH < 3.0, and solvent B was 10mM potassium diphosphate, 350mM potassium chloride, 25% ACN, pH < 3.0. The iTRAQ labeled peptides were diluted 1:4 in solvent A and applied to the SCX column. Chromatography was performed with a flow rate of 0.3 mL/min using the following gradient: 0-10 minutes, 0% solvent B; 10-60 minutes, 0%-100% solvent B; 60-65 minutes, 100% solvent B; 65-90 minutes, 0%-100% solvent A. For each high-performance liquid chromatography run, 24 of 27 fractions were collected at each peak and partially evaporated to remove ACN on a speed vacuum.
SCX sample clean-up and MALDI spotting
The fractions collected from the SCX fractionation were resolved in 5% ACN, 0.1% trifluoroacetic acid, pooled such that each pool contained 3 fractions, and desalted with Sep Pak C18 cartridges (Waters). For further separation, each peptide pool was dried and reconstituted in 10 μL 3% ACN, 0.2% formic acid, and 5 μL of each sample was loaded onto a reversed-phase column. Eluted peptides were mixed with MALDI matrix (3 mg/mL a-cyano-4-hydroxycinnamic acid, 0.1% trifluoroacetic acid, 70% ACN) and automatically spotted onto a MALDI plate (Applied Biosystems) using a Probot fraction collector. A total of 416 spots were collected from each high-performance liquid chromatography run.
MALDI-TOF/TOF measurement
MALDI plates were analyzed on a 4800 MALDI time of flight (TOF)/TOF system (Applied Biosystems) equipped with a Nd:YAG laser operating at 200 Hz. All mass spectra were acquired in positive reflector mode with data from 800 laser shots. The MS spectra were recalibrated internally based on the ion signal of neurotensin peptide (Sigma-Aldrich).
The following threshold criteria and settings were used for the acquisition of MS/MS spectra: mass range: 800-4000 Da; minimum signal-to-noise for MS/MS acquisition: 100; maximum number of peaks/spot: 8. Peptide collision-induced dissociation was performed at a collision energy of 1 kV and a collision gas pressure of approximately 2.5 × 10−6 Torr.
Data analysis
Acquired MS/MS spectra were interpreted using Mascot search algorithm version 2.1 embedded in the GPS (Global Proteome Server) Explorer software version 3.5 (Applied Biosystems). All searches were performed against a combined forward/reverse human (Homo sapiens) protein sequence database (fgcz_9606_d_20090511; 58 803 forward and 58 803 reverse sequences). Automatic normalization of quantitative data were performed to correct any experimental or systematic bias. The following search settings were used: maximum missed cleavages: 1; peptide mass tolerance: 25 ppm; MS/MS fragment tolerance: 0.25 Da; minimum ion score confidence interval for peptides: 90%. iTRAQ labeling at the N-terminal and lysine residues and blocking at the cysteine residues with methyl methanethiosulfonate (MMTS) were selected as fixed modifications. The false positive discovery rates (FDR) were calculated as a function of the Mascot total protein ion scores. At the lowest ion scores, after omitting all matches that were based on single peptide identification, the FDR was < 1% in all 3 experiments. Further data analysis was performed using Matlab 7.9 (MathWorks).
Gene-array experiments and data analysis
Total RNA was isolated using the RNeasy mini kit (QIAGEN), according to the manufacturer's instructions and including a DNase I digestion step (QIAGEN). Gene-expression profiling was performed by competitive dual-color hybridization of complementary RNA probes on human 4 × 44 K oligonucleotide microarray chips (Agilent Technologies), as described in our previous work.23 Before hybridization, we verified the integrity of each RNA sample on a RNA 6000 Nanochip (Bioanalyzer 2100 instrument; Agilent Technologies). High-quality RNA typically had an 18S:28S ribosomal RNA ratio > 1.5. cRNA probes were synthesized with the low-input fluorescence linear amplification kit protocol and spiked with control targets (Agilent Technologies). Equal quantities of Cy3- and Cy5-labeled probes were mixed, fragmented, and hybridized to the microarray slides. The arrays were scanned with a dual-laser microarray scanner and the raw data extracted with Feature Extraction software Version 9.5 (both Agilent Technologies). For statistical analysis, we used the Rosetta Resolver 7.2 Gene Expression Data Analysis System (Rosetta Biosoftware). Two-way analysis of variance (ANOVA) analysis was performed on the sequence level with specific parameters set as follows: ANOVA type: error-weighted, Benjamini-Hochberg FDR multiple test correction. Thresholds for sequence inclusion were set at a > 2-fold regulation, P < .01. Pair-wise analysis of changes in specific gene expression levels (ie, Dexa vs Dexa + Hb:Hp) was performed on the Unigene cluster level after combining experimental data of multiple replicates with the Rosetta Resolver in-built error algorithm. All gene array data are accessible at http://www.ebi.ac.uk/ebisearch/search.ebi?db = gene Expression&t = E-MEXP-2598.
Quantitative real-time RT-PCR
Real-time reverse transcription–polymerase chain reaction (RT-PCR) was performed on a Fast Real-Time PCR System Instrument (Applied Biosystems) using TaqMan reverse transcription and SYBR Green master mix PCR reagents (Applied Biosystems) as described previously.23 Gene-specific quantitative data were corrected for HPRT RNA abundance in the respective sample and are expressed as fold-expression relative to the untreated control. Sequences of the gene-specific primers can be found in supplemental Table 1 (available on the Blood Web site; see the Supplemental Materials link at the top of the online article).
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis and Western blot analysis and measurement of intracellular GSH and ferritin
Total cellular protein was extracted using CelLytic-M reagent (Sigma-Aldrich) supplemented with Complete Mini Protease Inhibitor (Roche Diagnostics). After 3 freeze-thaw cycles and sonication with a Branson Sonifier 250, cellular debris was removed by centrifugation at 16 000g for 15 minutes. Western blots were performed using standard techniques and the following primary antibodies: HO-1 (SPA-896; Stressgen), ferroportin (MTP11-A; Alpha Diagnostics), and transferrin receptor (MEM-189; Abcam). For detection, horseradish peroxidase–conjugated secondary antibodies (Amersham Biosciences) were used at a dilution of 1:10 000. Blots were developed with ECL Plus Western blotting detection reagent (Amersham Biosciences) and analyzed on a ChemiDoc XRS system with Quantity One 1-D Analysis software, Version 4.5.0 (Bio-Rad). Intracellular ferritin and reduced glutathione (GSH) were measured as described in our previous work.23
Ex vivo analysis of fluorescent Hb:Hp uptake in patient blood samples
Freshly drawn heparinized blood (100 μL) from routine blood samples of glucocorticoid-treated or -untreated patients was diluted with Gey balanced salt solution (1:1, Sigma-Aldrich). The diluted blood was incubated with 3 μg/mL Alexa 633-labeled Hb:Hp for 30 minutes in a 37°C water bath, and then the erythrocytes were lysed with EC-lysis solution (8.3 mg/mL ammonium chloride, 1 mg/mL potassium hydrogen carbonate, 0.037 mg/mL EDTA). EDTA was included in the lysis solution to prevent further Hb:Hp uptake by leukocytes and to remove bound, noningested ligand. After red blood cell lysis, the leukocytes were washed twice in PBS and subsequently stained with monoclonal antibodies against CD14 (fluorescein isothiocyanate) and CD64 (PE; Becton Dickinson). Cells were analyzed with a FACSCalibur (Becton Dickinson) equipped with a 488-nm argon laser and a 633-nm helium-neon laser. Data were analyzed using CellQuest (Becton Dickinson) and visualized with FlowJo 7 software (TreeStar). For analysis of cell surface expression of CD163, leukocytes were stained with an allophycocyanin-labeled anti-CD163 antibody (R&D Systems). Fluorescence micro-images were acquired with an Axioskop 2 epifluorescence microscope equipped with an oil-immersion 100×/1.3 objective lens, an AxioCam MR digital camera, and AxioVision software (Zeiss).
Results
LC-MALDI-MS/MS analysis identifies the Hb:Hp-CD163-HO1 pathway as a primary target of glucocorticoid activity in human monocytes
We investigated the effects of glucocorticoids on the global proteome composition of Hb-exposed monocytes. Figure 1 shows representative monocyte cell pellets at the end of an experiment. Monocytes that were pretreated with dexamethasone for 36 hours before Hb:Hp exposure clearly had a different appearance from control cells, and the red-brown color of these cells suggests that more intracellular Hb/heme accumulated in glucocorticoid-primed monocytes than in cells that were not pretreated. By combining isobaric tagging for relative and absolute quantification (iTRAQ) with LC-MALDI-MS/MS (liquid chromatography–matrixassisted laser desorption/ionization-mass spectrometry), we could identify and relatively quantify 1034 different monocyte proteins (461, 210, and 363 proteins in 3, 2, or 1 experiments, respectively) (supplemental Figure 1A). The identification of each protein was based on at least 2 peptide MS/MS matches. The calculated FDR was < 1% in all 3 experiments (supplemental Figure 1B). The experimental setup of the iTRAQ LC-MALDI-MS/MS experiments is shown in Figure 1. Figure 2 shows the 25 proteins that were most strongly increased or decreased after Hb:Hp exposure of dexamethasone-primed cells (red boxes). In this graph, each box indicates an individual protein detected in at least 2 of 3 experiments. The protein ratios for cells treated with Hb:Hp alone (ie, without dexamethasone pretreatment) are displayed as gray boxes. Therefore, the ratio difference between the red and gray signatures in Figure 2 points to a significant dexamethasone priming effect. From a functional perspective, we found that proteins functionally associated with Hb:Hp endocytosis and breakdown were most strongly enhanced in human monocytes pretreated with dexamethasone. A reproducible increase was observed in the intracellular concentrations of the 2 Hb subunits (α and β), Hp, HO-1, as well as H- and L-ferritin (heavy and light chains). In contrast, the functionally most significant iron importer, transferrin receptor (TfR), was the most strongly suppressed protein. In monocytes without dexamethasone pretreatment, Hb:Hp exposure resulted in a slight increase in the intracellular concentrations of Hb (α and β-globins), Hp, HO-1, and ferritin; however, the respective protein ratios of dexamethasone-primed monocytes were significantly greater than those of nonprimed cells. The complete LC-MALDI-MS/MS data are accessible in supplemental Excel File 1.

Experimental set-up for comparative monocyte proteome profiling. Monocytes were treated with or without dexamethasone (2.5 × 10−7M) for 36 hours. After this priming period, we added 500 μg/mL Hp (control cells) or 500 μg/mL Hb:Hp (1:1 molar ratio) for another 18 hours. At the end of each experiment, we observed a clearly visible difference in the appearance of cell pellets, with cells that were primed with glucocorticoid before exposure to Hb:Hp appearing more red-brown in color. Cells were then lysed, the nuclei removed by centrifugation, and the remaining cellular proteome processed for LC-MALDI-TOF/TOF analysis. Results of the comparative proteome analyses are shown in Figures 2 and 3, as indicated.
Experimental set-up for comparative monocyte proteome profiling. Monocytes were treated with or without dexamethasone (2.5 × 10−7M) for 36 hours. After this priming period, we added 500 μg/mL Hp (control cells) or 500 μg/mL Hb:Hp (1:1 molar ratio) for another 18 hours. At the end of each experiment, we observed a clearly visible difference in the appearance of cell pellets, with cells that were primed with glucocorticoid before exposure to Hb:Hp appearing more red-brown in color. Cells were then lysed, the nuclei removed by centrifugation, and the remaining cellular proteome processed for LC-MALDI-TOF/TOF analysis. Results of the comparative proteome analyses are shown in Figures 2 and 3, as indicated.
![Figure 2. Dexamethasone priming enhances CD163/HO-1–mediated Hb clearance and detoxification. Comparative proteome analyses of untreated and Hb:Hp-treated monocytes that were primed with dexamethasone (red signatures) or unprimed (black signatures). (A) All 1034 proteins were identified by more than 2 peptides. Each signature (bar) represents the maximum and minimum protein ratios (ie, [protein XYZ in Hb:Hp + dexamethasone cells]/[protein XYZ in control cells]). Proteins were sorted along the x-axis according to their expression level in monocytes treated with both dexamethasone and Hb:Hp. To allow direct comparison of the expression changes of each protein, the corresponding protein values for cells treated with Hb:Hp alone (ie, without dexamethasone priming) are shown at the same position on the x-axis. The difference between the 2 ratios (the space between the red and black symbols) can be attributed to the dexamethasone priming effect. (B) The top 25 up-regulated and top 25 down-regulated proteins from Figure 1A. A complete list of proteins, including identifications and scores, is provided in supplemental Table 2. Black/red signatures indicate proteins found in all 3 independent experiments. Gray/orange signatures indicate proteins identified in 2 experiments.](/view-large/figure/8004053/zh89991061250002.jpeg)
Dexamethasone priming enhances CD163/HO-1–mediated Hb clearance and detoxification. Comparative proteome analyses of untreated and Hb:Hp-treated monocytes that were primed with dexamethasone (red signatures) or unprimed (black signatures). (A) All 1034 proteins were identified by more than 2 peptides. Each signature (bar) represents the maximum and minimum protein ratios (ie, [protein XYZ in Hb:Hp + dexamethasone cells]/[protein XYZ in control cells]). Proteins were sorted along the x-axis according to their expression level in monocytes treated with both dexamethasone and Hb:Hp. To allow direct comparison of the expression changes of each protein, the corresponding protein values for cells treated with Hb:Hp alone (ie, without dexamethasone priming) are shown at the same position on the x-axis. The difference between the 2 ratios (the space between the red and black symbols) can be attributed to the dexamethasone priming effect. (B) The top 25 up-regulated and top 25 down-regulated proteins from Figure 1A. A complete list of proteins, including identifications and scores, is provided in supplemental Table 2. Black/red signatures indicate proteins found in all 3 independent experiments. Gray/orange signatures indicate proteins identified in 2 experiments.
Dexamethasone priming enhances CD163/HO-1–mediated Hb clearance and detoxification. Comparative proteome analyses of untreated and Hb:Hp-treated monocytes that were primed with dexamethasone (red signatures) or unprimed (black signatures). (A) All 1034 proteins were identified by more than 2 peptides. Each signature (bar) represents the maximum and minimum protein ratios (ie, [protein XYZ in Hb:Hp + dexamethasone cells]/[protein XYZ in control cells]). Proteins were sorted along the x-axis according to their expression level in monocytes treated with both dexamethasone and Hb:Hp. To allow direct comparison of the expression changes of each protein, the corresponding protein values for cells treated with Hb:Hp alone (ie, without dexamethasone priming) are shown at the same position on the x-axis. The difference between the 2 ratios (the space between the red and black symbols) can be attributed to the dexamethasone priming effect. (B) The top 25 up-regulated and top 25 down-regulated proteins from Figure 1A. A complete list of proteins, including identifications and scores, is provided in supplemental Table 2. Black/red signatures indicate proteins found in all 3 independent experiments. Gray/orange signatures indicate proteins identified in 2 experiments.
CD163 induction is a quantitatively major response of human monocytes to dexamethasone treatment
Dexamethasone pretreatment results in significant enhancement of Hb:Hp uptake and heme catabolism in Hb:Hp-exposed monocytes, suggesting that the Hb scavenger receptor CD163 could be an essential target of glucocorticoids. Figure 3 shows a correlation plot of 2 independent iTRAQ LC-MALDI-MS/MS experiments comparing the peptide composition of dexamethasone-treated and control monocytes. A total of 462 proteins were identified and quantified in both experiments, and, of these, CD163 was the seventh most strongly up-regulated protein in dexamethasone-treated monocytes. Immunohistochemistry and fluorescent Hb:Hp-uptake assays confirmed that dexamethasone-treated monocytes exhibited enhanced CD163 expression and highly increased Hb:Hp uptake capacity, respectively (Figure 3C). In agreement with the generally accepted catabolic activity of glucocorticoids, all ribosomal proteins identified by LC-MALDI-MS/MS were among the down-regulated protein matches. The prototypic M2/alternatively activated macrophage protein mannose receptor was also found among the top 10 up-regulated proteins. The complete LC-MALDI-MS/MS data are accessible in supplemental Excel File 2. Gene array analysis of the dexamethasone-induced gene expression pattern supports that the prominent CD163 induction occurs in agreement with a general M2 (alternatively activated) macrophage polarization. Supplemental Figure 2 shows a comparison of the RNA expression profiles of dexamethasone-treated and -nontreated human monocytes. M2 signature genes (ie, the interleukin-1 decoy-receptor, mannose receptor, and arginase) and/or genes known to be highly induced by glucocorticoids (ie, adenosine A3 receptor and FK506 binding protein) have been compiled from the literature and are highlighted alongside with down-regulated M1 signature genes such as the chemokines interleukin-8 and CXCL10 and indoleamine 2,3 dioxygenase.24-29

CD163 induction is a major effect of global glucocorticoid activity in monocytes. (A) iTRAQ MALDI-MS/MS analysis of the effects of glucocorticoid on the monocyte proteome. Monocytes were incubated for 36 hours with dexamethasone and then analyzed, as shown in Figure 1. A total of 1023 proteins were identified and relatively quantified (ratio of protein abundance for dexamethasone treated/control) based on at least 2 peptide matches. (B) Correlation plot of the 462 proteins identified in both independent experiments. Cyan dots represent ribosomal proteins; CD163 is shown in red. The prototypic M2 signature protein mannose receptor is highlighted in green. The complete list of up- and down-regulated proteins is given in supplemental Table 3. (C) Confirmation of CD163 up-regulation and increased Hb:Hp endocytosis in human monocytes after dexamethasone treatment. Monocytes were cultured on glass coverslips and stained with anti-CD163 monoclonal antibody (5C6FAT) and a secondary Alexa 568 goat anti-mouse antibody or incubated with 10 μg/mL Alexa594 Hb:Hp for 30 minutes. Images were acquired with a Zeiss epifluorescence microscope. Red indicates CD163 (right images) or internalized Hb:Hp (left images); blue shows DAPI (4′,6-diamidino-2-phenylindole)-stained nuclei.
CD163 induction is a major effect of global glucocorticoid activity in monocytes. (A) iTRAQ MALDI-MS/MS analysis of the effects of glucocorticoid on the monocyte proteome. Monocytes were incubated for 36 hours with dexamethasone and then analyzed, as shown in Figure 1. A total of 1023 proteins were identified and relatively quantified (ratio of protein abundance for dexamethasone treated/control) based on at least 2 peptide matches. (B) Correlation plot of the 462 proteins identified in both independent experiments. Cyan dots represent ribosomal proteins; CD163 is shown in red. The prototypic M2 signature protein mannose receptor is highlighted in green. The complete list of up- and down-regulated proteins is given in supplemental Table 3. (C) Confirmation of CD163 up-regulation and increased Hb:Hp endocytosis in human monocytes after dexamethasone treatment. Monocytes were cultured on glass coverslips and stained with anti-CD163 monoclonal antibody (5C6FAT) and a secondary Alexa 568 goat anti-mouse antibody or incubated with 10 μg/mL Alexa594 Hb:Hp for 30 minutes. Images were acquired with a Zeiss epifluorescence microscope. Red indicates CD163 (right images) or internalized Hb:Hp (left images); blue shows DAPI (4′,6-diamidino-2-phenylindole)-stained nuclei.
Simultaneous glucocorticoid treatment and Hb:Hp exposure induces antioxidant and iron recycling genes
After establishing that dexamethasone promotes a monocyte phenotype with activated Hb clearance capacity, we performed 2 different gene array experiments to analyze the interactive effects of concurrent glucocorticoid treatment and Hb:Hp exposure on the monocyte transcriptome. This simultaneous exposure of monocytes to extracellular Hb and glucocorticoids is similar to that experienced during treatment of hemolysis. The experimental setup of these studies is shown in Figure 4. The factorial experiment involved pair-wise comparisons of a hybridization sample from control cells (gray box) with hybridization samples from native or dexamethasone pretreated monocytes that had been exposed to Hb:Hp for 0, 8, and 18 hours. In this experiment, dexamethasone and Hb:Hp treatment were considered independent factors in the 2-way ANOVA. Only changes with a significant interactive component between Hb:Hp and dexamethasone were considered for further analysis. This analysis yielded 905 sequences. Supplemental Figure 2A-B illustrates the results of the 2-way ANOVA with exemplary quantitative PCR confirmations. We also explored the RNA expression changes that are induced by Hb:Hp in dexamethasone pretreated monocytes by direct pair-wise comparison. Using this approach, 383 genes that were induced > 1.5-fold by Hb:Hp in dexamethasone-primed monocytes were selected for further analysis. The combination of these 2 selection thresholds identified 47 genes that were significantly coregulated by dexamethasone priming and Hb:Hp exposure and were induced > 1.5-fold in dexamethasone-primed cells. These “genes of interest” are listed in Table 1.

Experimental setup for gene array analysis of the effect of dexamethasone priming on Hb:Hp-induced gene expression. Monocytes were cultured in the presence or absence of dexamethasone (2.5 × 10−7M) for 36 hours (priming phase). After this priming period, 500 μg/mL purified Hp or Hb:Hp (1:1 molar ratio) was added. After incubation for an additional 0, 8, or 18 hours, RNA was extracted, and samples were analyzed with Agilent whole genome 44K arrays. Two different array experiments were performed to achieve a high stringency for the selection of genes of interest. (1) A factorial experiment (blue) to identify sequences with a significant (P < .01) interactive component of dexamethasone priming and Hb:Hp treatment was performed. For factorial analysis, each treatment sample was analyzed against a reference (untreated) sample from the same experiment (gray box). Ratio-data of 4 independent experiments were analyzed by 2-way ANOVA considering the factors of dexamethasone treatment and time of Hb:Hp exposure. The 905 sequences that showed a significant interactive component (P < .01) were considered for further analysis. (2) A pair-wise experiment (green) was performed for direct identification of genes that were significantly (P < .01) induced/suppressed > 1.5-fold when dexamethasone-primed cells were exposed to Hb:Hp. The 383 genes fulfilling this second threshold were considered for further analysis. Combination of the 2 criteria identified 47 genes of interest, which are shown in Table 1.
Experimental setup for gene array analysis of the effect of dexamethasone priming on Hb:Hp-induced gene expression. Monocytes were cultured in the presence or absence of dexamethasone (2.5 × 10−7M) for 36 hours (priming phase). After this priming period, 500 μg/mL purified Hp or Hb:Hp (1:1 molar ratio) was added. After incubation for an additional 0, 8, or 18 hours, RNA was extracted, and samples were analyzed with Agilent whole genome 44K arrays. Two different array experiments were performed to achieve a high stringency for the selection of genes of interest. (1) A factorial experiment (blue) to identify sequences with a significant (P < .01) interactive component of dexamethasone priming and Hb:Hp treatment was performed. For factorial analysis, each treatment sample was analyzed against a reference (untreated) sample from the same experiment (gray box). Ratio-data of 4 independent experiments were analyzed by 2-way ANOVA considering the factors of dexamethasone treatment and time of Hb:Hp exposure. The 905 sequences that showed a significant interactive component (P < .01) were considered for further analysis. (2) A pair-wise experiment (green) was performed for direct identification of genes that were significantly (P < .01) induced/suppressed > 1.5-fold when dexamethasone-primed cells were exposed to Hb:Hp. The 383 genes fulfilling this second threshold were considered for further analysis. Combination of the 2 criteria identified 47 genes of interest, which are shown in Table 1.
Interactive effects of Dexamethasone and Hb:Hp-on monocyte gene expression
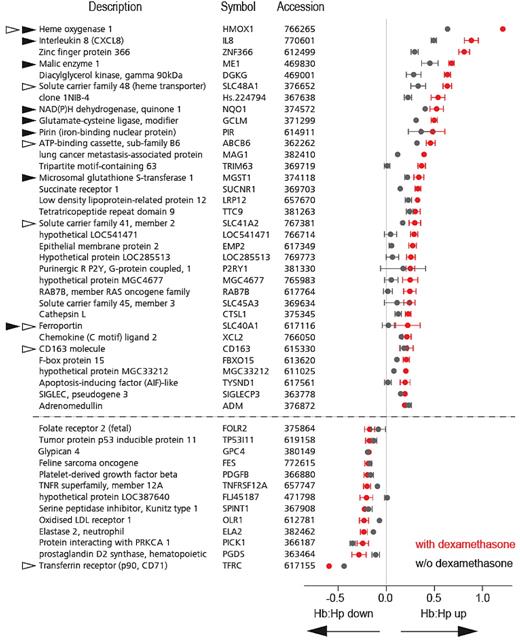
The table shows the 47 genes identified by the gene array analysis described in Figure 4. These genes were significantly co-regulated by dexamethasone priming and Hb:Hp exposure and were induced >1.5-fold in dexamethasone-primed cells. The graph on the right shows the corresponding gene expression changes (log ratio; mean ± error, n=4) induced by Hb:Hp in either dexamethasone pretreated (red) or nontreated (black) monocytes. Note that almost all genes on this list exhibit greater induction or suppression in monocytes pretreated with dexamethasone. Black arrows indicate genes known to be regulated by the Nrf-2–Maf/MARE signaling pathway. White arrows indicate genes with functions related to Hb, heme, or iron metabolism.
We manually checked the available literature to identify known functions and regulatory elements of the 47 transcripts shown in Table 1 and tried to classify them into functional groups. It was possible to assign 12 of the 34 up-regulated genes (32.3%) into 2 functional groups: (1) conserved antioxidant response (7 genes) and (2) Hb/heme/iron metabolism (6 genes; indicated in Table 1). For each of the genes assigned to Group 1, we found published evidence for transcriptional control by the major oxidative stress-response regulator nuclear factor E2-related factor (Nrf-2). Therefore, dexamethasone treatment appears to shift the differentiation of Hb:Hp-exposed monocytes into an antioxidant and heme-iron recycling phenotype.
We next performed an independent series of experiments to confirm that dexamethasone supports key antioxidant and heme-iron homeostasis genes/proteins during Hb exposure of monocytes. In these studies, we used 50% autologous serum as a substitute for the purified Hp used in the discovery experiments. The serum was used in combination with 2 different Hbs: a highly purified HbA0 and an αα-cross-linked tetrameric Hb that can be endocytosed efficiently by CD163 via an Hp-independent mechanism.30 In both cases, dexamethasone pretreatment significantly enhanced induction of the rate-limiting GSH synthesis enzyme glutamate-cysteine ligase (GCLM) by Hb, resulting in a measurable increase in intracellular reduced GSH (Figure 5A). In addition, dexamethasone pretreatment augmented the Hb-induced increase in mRNA and protein for the heme breakdown enzyme HO-1 and intracellular ferritin levels (Figure 5B). The ferritin measured by this chemiluminescent assay primarily represents L-ferritin, which is the predominant form of ferritin in plasma. Together with the increased levels of L- and H-ferritin peptides found by mass spectrometry, this finding supports an enhanced iron load of dexamethasone/Hb:Hp-treated monocytes. Furthermore, in monocytes pretreated with glucocorticoid and exposed to Hb, the transferrin receptor was suppressed, while mRNA and protein levels of the principle iron exporter ferroportin were increased (Figure 5C). These data strongly point to a profound shift in antioxidant and iron metabolism when Hb-exposed monocytes are treated with glucocorticoids. These adaptive changes are almost completely inhibited by inflammatory monocyte stimulation with endotoxin (supplemental Figure 4).

RNA and protein changes of core antioxidant and iron metabolism genes responsive to the interactive effects of dexamethasone and Hb. Monocytes were primed with dexamethasone for 36 hours (■) or left untreated ( ) and then incubated with medium containing 50% autologous serum (c), 400 μg/mL HbA0 (Hb), or 400 μg/mL tetramer-stabilized α-α crosslinked Hb (αα). Cells were treated with Hb for 12 hours before mRNA analysis or incubated for 24 hours for determination of intracellular reduced GSH and ferritin or Western blot analysis. (A) Changes in mRNA expression of GCLM (left) and level of Hb-induced intracellular GSH (right) in dexamethasone-primed monocytes. (B) mRNA and protein expression levels of heme oxygenase (HO-1) (left and lower) and intracellular ferritin levels (right). (C) mRNA and protein levels of the transferrin receptor (TfR1) and the iron exporter ferroportin (Fpn1). All quantitative results represent mean ± SEM of at least 3 independent experiments.
) and then incubated with medium containing 50% autologous serum (c), 400 μg/mL HbA0 (Hb), or 400 μg/mL tetramer-stabilized α-α crosslinked Hb (αα). Cells were treated with Hb for 12 hours before mRNA analysis or incubated for 24 hours for determination of intracellular reduced GSH and ferritin or Western blot analysis. (A) Changes in mRNA expression of GCLM (left) and level of Hb-induced intracellular GSH (right) in dexamethasone-primed monocytes. (B) mRNA and protein expression levels of heme oxygenase (HO-1) (left and lower) and intracellular ferritin levels (right). (C) mRNA and protein levels of the transferrin receptor (TfR1) and the iron exporter ferroportin (Fpn1). All quantitative results represent mean ± SEM of at least 3 independent experiments.
RNA and protein changes of core antioxidant and iron metabolism genes responsive to the interactive effects of dexamethasone and Hb. Monocytes were primed with dexamethasone for 36 hours (■) or left untreated () and then incubated with medium containing 50% autologous serum (c), 400 μg/mL HbA0 (Hb), or 400 μg/mL tetramer-stabilized α-α crosslinked Hb (αα). Cells were treated with Hb for 12 hours before mRNA analysis or incubated for 24 hours for determination of intracellular reduced GSH and ferritin or Western blot analysis. (A) Changes in mRNA expression of GCLM (left) and level of Hb-induced intracellular GSH (right) in dexamethasone-primed monocytes. (B) mRNA and protein expression levels of heme oxygenase (HO-1) (left and lower) and intracellular ferritin levels (right). (C) mRNA and protein levels of the transferrin receptor (TfR1) and the iron exporter ferroportin (Fpn1). All quantitative results represent mean ± SEM of at least 3 independent experiments.
Monocytes from patients on glucocorticoid pulse therapy show enhanced Hb:Hp-uptake capacity
Next, we established whether clinically relevant glucocorticoid treatment protocols could enhance Hb:Hp-uptake capacity and potential downstream metabolic pathways in vivo. We performed an ex vivo whole blood Hb:Hp-uptake assay on adult patients with nonmalignant hematologic or autoimmune diseases or transplant rejection who were treated with glucocorticoid pulse therapies. All samples were analyzed within 3 days after the start of glucocorticoid pulse therapy, which involved an average treatment dose of 886 mg/d methylprednisolone equivalent per patient. This simple assay confirmed that among the major peripheral blood leukocyte populations, only CD14+/CD163+ monocytes could endocytose fluorescent Hb:Hp (Figure 6A). While the negligible uptake of Hb:Hp by lymphocytes and granulocytes was not enhanced on glucocorticoid treatment, we found that CD14+/CD163+ monocytes from patients treated with glucocorticoids showed a significantly enhanced uptake of Hb:Hp (Figure 6B).

Glucocorticoid pulse therapy enhances peripheral blood monocyte Hb:Hp-uptake capacity in vivo. Fluorescent Hb:Hp633-uptake capacity of peripheral blood leukocytes was measured ex vivo in routine blood samples from patients without glucocorticoid treatment (control) and from patients after glucocorticoid pulse therapy (GC). (A) A distinct cell population (red frame) with Hb:Hp633-uptake capacity can be identified in the plot shown in the left panel. The CD14high/CD163high phenotype (red) of these cells (right) identifies the cell population as monocytes expressing the Hb scavenger receptor. Black indicates granulocytes, and gray indicates lymphocytes. (B) The mean FL4 (Hb:Hp633) fluorescence of each cell population was extracted in the patient samples as shown in A. The CD14high/CD163high monocytes from individuals treated with a glucocorticoid pulse (n = 14) showed a significantly (P < .001) increased Hb:Hp-uptake capacity compared with nontreated individuals (n = 11).
Glucocorticoid pulse therapy enhances peripheral blood monocyte Hb:Hp-uptake capacity in vivo. Fluorescent Hb:Hp633-uptake capacity of peripheral blood leukocytes was measured ex vivo in routine blood samples from patients without glucocorticoid treatment (control) and from patients after glucocorticoid pulse therapy (GC). (A) A distinct cell population (red frame) with Hb:Hp633-uptake capacity can be identified in the plot shown in the left panel. The CD14high/CD163high phenotype (red) of these cells (right) identifies the cell population as monocytes expressing the Hb scavenger receptor. Black indicates granulocytes, and gray indicates lymphocytes. (B) The mean FL4 (Hb:Hp633) fluorescence of each cell population was extracted in the patient samples as shown in A. The CD14high/CD163high monocytes from individuals treated with a glucocorticoid pulse (n = 14) showed a significantly (P < .001) increased Hb:Hp-uptake capacity compared with nontreated individuals (n = 11).
Discussion
Our overall focus is to explore novel activities of glucocorticoids that could support adaptive responses to extracellular Hb exposure independent of common anti-inflammatory and immunosuppressive mechanisms. Although glucocorticoid regulation has been reported for multiple fragments of the mutually interactive Hb-clearance, heme breakdown, and iron and antioxidant metabolism, we provide the first “global” analysis of the orchestrated response of monocytes to combined glucocorticoid and Hb exposure. Our data provide evidence that the adaptive response to extracellular Hb should be considered a primary target of glucocorticoid activity. We specifically found that (1) the most prominent in vitro and in vivo changes observed in glucocorticoid-treated monocytes were enhanced expression of the Hb scavenger receptor CD163, increased Hb:Hp uptake, and increased heme-degradation capacity and (2) the interactive effects of dexamethasone and Hb shift the monocyte phenotype toward enhanced iron-recycling and antioxidant homeostasis.
iTRAQ-based mass spectrometry and gene array analysis indicated that enhanced expression of the CD163 scavenger receptor was one of the most significant noncatabolic impacts of glucocorticoid treatment of monocytes. The enhanced CD163 expression was found to be in agreement with a general M2 transcriptome polarization, which characterizes an anti-inflammatory macrophage phenotype considered to be involved in the down-regulation of acute inflammation and wound healing.24-29 Upon concurrent treatment with dexamethasone and Hb:Hp, the monocyte proteome shifted toward a signature consistent with enhanced endocytosis of Hb:Hp, enhanced heme breakdown by HO-1 and storage of Hb-derived iron within ferritin, and cellular exclusion of transferrin iron. These changes reflect a major gain-of-function effect of glucocorticoids that supports a very specific homeostatic role of monocytes.
Our transcriptome analysis confirmed and considerably expanded the idea that glucocorticoids prime the monocyte response to Hb and reshape the heme, iron, and antioxidant metabolic pathways during Hb exposure. Extracellular Hb can induce peroxidative stress, which eventually damages susceptible biomolecules, whole cells, and tissues. Therefore, enhancement of antioxidative metabolisms might be an essential adaptive response to Hb exposure. In the current work, we found that glucocorticoid treatment significantly enhanced the Hb-induced levels of the major intracellular antioxidant GSH via super-induction of the rate-limiting enzyme GCLM. Dexamethasone also enhanced the induction of other antioxidant enzymes by Hb:Hp, including malic enzyme (ME1), NAD(P)H:quinone oxidoreductase (NQO1), and GSH S-transferase (GST1). ME1 fuels a core metabolic pathway to reconstitute nicotinamide adenine dinucleotide phosphate-oxidase (NADPH) and therefore is essential for maintaining a high level of reduced GSH. Glucocorticoid priming also supported pathways that specifically mitigate heme- and iron-associated toxicity. Both gene array analysis and mass spectrometry identified HO-1 as the enzyme that was most strongly induced by Hb in dexamethasone-primed monocytes. The reaction products of this enzyme, which include carbon monoxide, bilirubin, and ferritin, specifically attenuate vaso-occlusion and inflammation in sickle cell mice.10,31
In monocytes with a high level of CD163 expression, Hb exposure is likely to challenge cellular iron homeostasis through a massive intracellular release of heme iron. In this situation, the cellular iron equilibrium could eventually be kept in balance by limiting acquisition of nonheme iron via down-regulation of the transferrin-receptor and concurrently enhancing cellular iron export by up-regulation of the divalent iron exporters SLC40A1 (ferroportin) and SLC41A2 (a low affinity iron exporter).32,33 In addition, excess iron can be redistributed to newly synthesized ferritin. These changes may limit potential intracellular heme iron toxicity. However, the shift of iron pathways toward enhanced export and suppression of transferrin-iron import would also support delivery of Hb-iron from monocytes/macrophages to the erythropoetic compartment to enhance the iron supply during hemolysis (eg, recycling).34 Glucocorticoids also appear to alter the intracellular heme distribution through up-regulation of ABCB6 and SLC48A1 transcripts that encode 2 heme transporters localized in lysosomal membranes.35,36 Similar to the heme carrier protein-1 that we have shown recently to be induced by glucocorticoid activity in macrophages, these additional transporters may support enhanced heme translocation from the lysosome (where Hb is degraded) to the cytoplasmic compartment (where heme can be decomposed by HO-1).37
Among the dexamethasone/Hb-regulated transcripts, we detected a striking overrepresentation of genes known to be responsive to activated Nrf-2 (Table 1). The transcription factor Nrf-2 interacts with small Maf proteins to activate Maf recognition elements (MARE) such as those found within the antioxidant response elements. Therefore, Nrf-2 is considered a central regulator of the antioxidant response.38 This antioxidant response induces a group of essential defense genes including HO-1, ME1, NQO1, GCLM, and GST1, all of which were targets of combined glucocorticoid/Hb activity in our study. In addition, activation of the Nrf-2-Maf/MARE pathway is also critically involved in the regulation of macrophage heme-iron recycling through transcriptional induction of ferroportin.39 We previously established that CD163-mediated Hb-heme internalization was the only signal required to induce high levels of HO-1 expression in CD163+ HEK293 cells. Heme and other porphyrins can directly stimulate Nrf-2-Maf/MARE-dependent transactivation through interaction with key control elements of the signaling pathway. When the intracellular heme level is low, the actin-associated protein Keap-1 inactivates Nrf-2 within the cytoplasm.38 Conversely, high intracellular heme levels inhibit the transcriptional repressor activity of Bach1 and thus directly stimulate Maf/MARE dependent transcription.40
Based on these known regulatory mechanisms, we propose the following model to explain the transcriptional and protein expression events observed in our experiments (summarized in Figure 7): Glucocorticoids stimulate monocyte CD163 expression (priming effect) and thereby enhance endocytosis of Hb:Hp complexes during hemolysis. Intracellular Hb degradation and heme release activate the Nrf-2-Maf/MARE pathway, which promotes expression of HO-1, antioxidative enzymes (GCLM, GST1, NQO1, and ME1), and the iron exporter ferroportin. The release of free iron from heme adds a third (posttranscriptional) level of regulation driven by the iron regulatory protein/iron responsive element. Activated iron regulatory protein/iron responsive element stimulates ferritin and ferroportin synthesis while suppressing transferrin receptor synthesis.41 The final result is an antioxidant monocyte phenotype with a high capacity to recycle Hb-derived iron during hemolysis.
![Figure 7. Schematic illustration of the glucocorticoid effect on monocytes during extracellular Hb exposure. Native circulating blood monocytes have a moderate capacity to clear extracellular Hb. Glucocorticoids induce CD163 expression and stimulate receptor mediated Hb:Hp endocytosis. The resulting increase in intracellular heme leads to activation of the Nrf-2-Maf/MARE signaling pathway with subsequent enhancement of HO-1, ferroportin, and antioxidative genes (malic enzyme [ME]; NAD(P)H dehydrogenase quinone 1 [NQO1]; GCLM). Free iron that is released during HO-1–mediated heme degradation can activate the iron-regulated protein (IRP)/iron-responsive element (IRE) leading to increased synthesis of ferritin, suppression of the transferrin receptor (TfR), and increased expression of iron exporter ferroportin (Fpn).](/view-large/figure/8004074/zh89991061250007.jpeg)
Schematic illustration of the glucocorticoid effect on monocytes during extracellular Hb exposure. Native circulating blood monocytes have a moderate capacity to clear extracellular Hb. Glucocorticoids induce CD163 expression and stimulate receptor mediated Hb:Hp endocytosis. The resulting increase in intracellular heme leads to activation of the Nrf-2-Maf/MARE signaling pathway with subsequent enhancement of HO-1, ferroportin, and antioxidative genes (malic enzyme [ME]; NAD(P)H dehydrogenase quinone 1 [NQO1]; GCLM). Free iron that is released during HO-1–mediated heme degradation can activate the iron-regulated protein (IRP)/iron-responsive element (IRE) leading to increased synthesis of ferritin, suppression of the transferrin receptor (TfR), and increased expression of iron exporter ferroportin (Fpn).
Schematic illustration of the glucocorticoid effect on monocytes during extracellular Hb exposure. Native circulating blood monocytes have a moderate capacity to clear extracellular Hb. Glucocorticoids induce CD163 expression and stimulate receptor mediated Hb:Hp endocytosis. The resulting increase in intracellular heme leads to activation of the Nrf-2-Maf/MARE signaling pathway with subsequent enhancement of HO-1, ferroportin, and antioxidative genes (malic enzyme [ME]; NAD(P)H dehydrogenase quinone 1 [NQO1]; GCLM). Free iron that is released during HO-1–mediated heme degradation can activate the iron-regulated protein (IRP)/iron-responsive element (IRE) leading to increased synthesis of ferritin, suppression of the transferrin receptor (TfR), and increased expression of iron exporter ferroportin (Fpn).
Macrophage Hb, iron, and antioxidant metabolisms are highly sensitive to inflammation and oxidative stress. Reduced cellular iron export with subsequent excess storage of iron in inflammatory activated macrophages is one of the causes of anemia of chronic disease.42 When monocytes and macrophages are stimulated by lipopolysaccharide and other toll-like receptor ligands or when exposed to oxidative stress mediators, CD163 is shed from the cell surface, leading to a reduction in Hb:Hp endocytic capacity.17,43,44 In agreement with these observations, Hb:Hp-stimulated HO-1 and ferroportin expression were both dramatically reduced when the dexamethasone pretreated monocytes were activated by lipopolysaccharide immediately before Hb:Hp exposure.
To investigate the activity of glucocorticoids in vivo, we examined the Hb:Hp-uptake capacity of peripheral blood leukocytes from patients treated with pulse glucocorticoid therapy and observed an approximately 4-fold increase in peripheral blood monocyte Hb:Hp-uptake capacity in these patients. These findings strongly suggest that the basic mechanisms underlying the glucocorticoid response are the same in vitro and in vivo. A limitation of our ex vivo study is that we could measure only the Hb-uptake capacity of peripheral blood monocytes in glucocorticoid treated patients but not of tissue macrophages. Tissue macrophages express high levels of the Hb-scavenger receptor, and the glucocortioid inducing effect on CD163 has been demonstrated in different in vitro macrophage cell culture models using peripheral blood monocyte derived macrophages as well as mouse peritoneal macrophages.18,21,45 It is therefore possible that a synergistic effect of glucocorticoids on monocytes and tissue macrophages could enhance systemic Hb clearance in vivo. We have previously shown that CD163 mRNA expression in peripheral blood monocytes rapidly returns to baseline levels after glucocorticoid withdrawal, and it remains to be established whether rapid loss of the Hb-adapted monocytes could relate to the frequent rebound disease activity that has been observed after glucocorticoid pulse therapies of sickle cell disease complications.46
Why is the adaptive monocyte response to Hb so responsive to glucocorticoids? It has been shown experimentally that even stress concentrations of cortisone can increase CD163 expression, and monocytes with significantly enhanced CD163 have been detected after infusion of endotoxin in healthy persons as well as in patients with severe burn injury.47-49 Therefore, it might be speculated that the stress-inducible Hb scavenger system has developed under the evolutionary pressure of infectious hemolytic diseases such as malaria. In these conditions, activation of the endogenous stress response might support monocyte/macrophage adaptation to extracellular Hb.50
In conclusion, we have established that glucocorticoid treatment shifts the monocyte phenotype toward enhanced Hb uptake and heme breakdown. In conjunction with downstream adaptive transcriptional and translational responses, this major noncatabolic activity of glucocorticoids may be essential for the protection against the adverse effects of extracellular Hb.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported by the Swiss National Science Foundation (grant 31-120658), the Fonds für Akademische Nachwuchsförderung (FAN), and the Helmut Horten Foundation.
Authorship
Contribution: F.V. and C.A.S. performed experiments, analyzed data, and wrote the paper; T.K. and P.G. performed mass spectrometry measurements and data analysis; E.D. analyzed data; and G.S. and D.J.S. designed the study, analyzed data, and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Dominik J. Schaer, Internal Medicine, University Hospital, CH-8091 Zurich, Switzerland; e-mail: dominik.schaer@usz.ch.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal