

Abstract
Microbes as well as immune complexes and other continuously generated inflammatory particles are efficiently removed from the human circulation by red blood cells (RBCs) through a process called immune-adherence clearance. During this process, RBCs use complement receptor 1 (CR1, CD35) to bind circulating complement-opsonized particles and transfer them to resident macrophages in the liver and spleen for removal. We here show that ligation of RBC CR1 by antibody and complement-opsonized particles induces a transient Ca++ influx that is proportional to the RBC CR1 levels and is inhibited by T1E3 pAb, a specific inhibitor of TRPC1 channels. The CR1-elicited RBC Ca++ influx is accompanied by an increase in RBC membrane deformability that positively correlates with the number of preexisting CR1 molecules on RBC membranes. Biochemically, ligation of RBC CR1 causes a significant increase in phosphorylation levels of β-spectrin that is inhibited by preincubation of RBCs with DMAT, a specific casein kinase II inhibitor. We hypothesize that the CR1-dependent increase in membrane deformability could be relevant for facilitating the transfer of CR1-bound particles from the RBCs to the hepatic and splenic phagocytes.
Introduction
In primates, in contrast to other vertebrates, clearing the intravascular space of complement-opsonized inflammatory particles (eg, microbes and immune complexes) is mediated by circulating red blood cells (RBCs) using complement receptor 1 (CR1, CD35).1,2 During this process, known as immune-adherence clearance, RBCs immobilize complement-tagged particles and transport them to the liver and spleen where resident macrophages remove the complement-tagged particles and leave the RBCs intact. Immune-adherence clearance acts as a “buffer system,” preventing deposition of circulating immune complexes in susceptible organs, such as the kidney, and preventing activation of circulating leukocytes by inflammatory particles.3,4 We and others have also shown that CR1-mediated immune-adherence promotes more efficient phagocytosis and intracellular killing of complement-opsonized pathogens compared with opsonized pathogens that are free-floating in plasma and not RBC-bound.5,6
We have previously found that, in circulating human RBCs, CR1 is disperse in RBC plasma membranes, and, after ligation by immune particles, interacts with Fas-associated phosphatase-1 and rearranges into large clusters.7 Beneath the plasma membrane of RBCs, the spectrin cytoskeleton defines a series of “corrals” that are critical for maintaining RBC shape and deformability and for regulating the range and magnitude of lateral diffusion of most transmembrane proteins.8 The mechanical attributes of the spectrin meshwork depend critically on the transient phosphorylation of β-spectrin, adducin, and protein 4.1R.9-11 Therefore, we hypothesized that ligation-mediated CR1 clustering is an active process with CR1 directly affecting the phosphorylation status of cytoskeletal proteins and thus the mechanical properties of RBCs. We here report that, in human RBCs, CR1 ligation induces a transient Ca++ influx that depends on stretch-activated transient receptor potential channel-1 (TRPC-1). In addition, CR1 ligation and Ca++ influx promote phosphorylation of the cytoskeletal proteins, α-adducin and β-spectrin, which correlates with increased membrane deformability. Our study identifies CR1 ligation as an important event affecting RBC membrane deformability, which in itself could have an important role during the immune-adherence clearance process.
Methods
Antibodies and reagents
Antibodies (Abs) were obtained as follows: anti-CR1 monoclonal Abs (mAb): 1F11 (gift of Henry Marsh, Celldex Therapeutics, Needham, MA), YZ-1,12 and 2B11,13 rabbit polyclonal anti-CR1,2 nonimmune immunoglobulin G1 (IgG1; BD Biosciences); anti-TRPC1 rabbit polyclonal (Santa Cruz Biotechnology); anti-TRPC1, T1E3 (gift of Yao Xiaoqiang, University of Hong Kong), anti-TRPC1 rabbit monoclonal anti-actin, anti-CD47, anti-adducin, anti-phospho-adducin (serine 726), anti-phospho serine/threonine mAbs, and anti–human glycophorin C (GPC) mAb (BRIC10; International Blood Group Reference Laboratory; Abcam). Secondary Abs included: AlexaFluor488 goat anti–mouse IgG, AlexaFluor488 goat anti–rabbit IgG, AlexaFluor594 goat anti–rabbit IgG “highly cross absorbed,” and AlexaFluor594 goat anti–mouse IgG “highly cross absorbed” (Invitrogen); horseradish peroxidase (HRP)-goat anti–mouse IgG, HRP-donkey anti–goat IgG, and HRP-donkey anti–rabbit IgG (Jackson ImmunoResearch Laboratories), GsMTx-4 (Peptide Institute). Reagents were obtained as follows: Fluo-4-AM, eosin 5 maleimide (Invitrogen); IgG-free bovine serum albumin (BSA; Jackson ImmunoResearch Laboratories); inhibitors for casein kinase I, D4476, and casein kinase II, 2-dimethylamino-4,5,6,7-tetrabromo-1H-benzimidazole (DMAT; EMD Chemicals); phorbol 12-myristate 13-acetate (PMA), 2-(N-morpholino) ethanesulfonic acid (MES), and 2-aminoethoxydiphenyl borate (2-APB; Sigma-Aldrich).
Analysis of RBC calcium influx
RBCs (108) were preloaded with Fluo-4 AM for 15 minutes at room temperature (RT), washed, and resuspended in Hank balanced salt solution (HBSS) with Ca++ and Mg++. RBCs were incubated at RT for an additional 10 minutes and washed once to remove any uncleaved Fluo-4 AM. Due to the ATP-depleting effect of the acetoxymethyl group, all experiments were performed within 1 hour from Fluo-4 AM loading. Fluorescence levels of RBCs were acquired for 15 seconds using a LSRII flow cytometer (Becton Dickinson) to establish a baseline for intracellular RBC Ca++ concentration. Control or anti-CR1 Ab were then added to the RBCs, mixed briefly, and RBC fluorescence intensity was recorded for 2 minutes. Data were exported as FSC 3.0 files without the time dimension parameter and analyzed using the kinetic module of FlowJo 9.0.1 (TreeStar). To measure saturated concentration of intracellular Ca++, Fluo-4-loaded RBCs were incubated with 5μM ionomycin. Intracellular Ca++ concentration after CR1 ligation was calculated using the following formula:
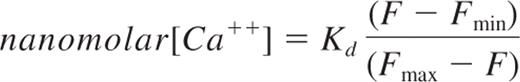
where Kd is the dissociation constant for Ca++-bound Fluo-4 (around 345nM)14 ; Fmax, the maximum intensity after ionomycin treatment; Fmin, the intensity of Fluo-4 loaded RBCs in the absence of Ca++; and F, the intensity of Fluo-4 after CR1 ligation.
Western blotting analysis
RBCs (2 μL packed cells) incubated with isotype control or anti-CR1 YZ-1 mAb (10 μg/mL) were lysed in 100 μL 1 × reducing-loading buffer and boiled for 4 minutes. Samples were run on 10% Tris-HCl gels (Invitrogen) and transferred to nitrocellulose paper (Pierce). Membranes were then incubated with Ab as noted in Figure legends, followed by appropriate HRP-conjugated secondary Ab, and developed using LAS 4000 imaging system (FujiFilm).
Assay of RBC membrane deformability using laser optical tweezers (LOT)
Anti-GPC mAb was covalently attached to 1-μm polystyrene beads with carboxyl surfaces (Polysciences). Beads (20 μL) were washed twice in 1 mL 20mM MES buffer, pH 6.1. The carboxyl groups on the beads were sensitized using 100mM N-hydroxysuccinimide and 0.4% 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride for 20 minutes at RT. Mouse anti-GPC antibody (10 μL of a 5 μg/mL solution) was added to the bead suspension and the mixture was incubated with constant shaking for 2 hours at RT. BSA was then added to 1% (final concentration), and the mixture was incubated for an additional 15 minutes. The conjugation reaction was terminated by adding ethanolamine (100mM final concentration) for another 15 minutes. The mixture was then washed twice in HBSS with 0.1% BSA. The conjugated beads were stored at 4°C and used within 1 week.
Anti-GPC-conjugated polystyrene beads were mixed with RBCs in HBSS with 0.5% BSA and placed between 2 glass coverslips to make a 30-μm–thick sample. Most of the RBCs attached to the glass by a small area on their rim. LOT (stiffness 7.7 10−5N/m, calibrated using harmonic potential approximation applied to the motion of an unattached trapped bead in solution) was used to trap a bead from the solution and bring the trapped bead into contact with a RBC for 5 seconds, which usually resulted in strong attachment. A computer-controlled procedure translated the trapped bead 2 μm at a rate of 20 μm/min extending the RBC into an elliptical shape, and then switched off the LOT, which released the bead and the pulled RBC recovered its circular (discocytic) shape. The time required for the shape recovery was calculated based on time-lapse images recorded at 30 frames/s. The first 0.5 seconds of each trajectory was fitted to an exponential with recovery time constant tc. This characteristic time for relaxation is given by tc = η/μ, where η is the viscosity of the RBC membrane, and μ is the shear elastic modulus of the membrane.15
Differences between the control and various experimental conditions were compared using the nonparametric one-way analysis of variance (ANOVA) followed by Tukey-Kramer multiple comparison procedure performed using MATLAB (MathWorks) and P values < .05 were considered statistically significant.
Assay of RBC deformability using a filtration device
To test the ability of RBCs to undergo capillary-like deformations, we fabricated a filter comprising an array of posts with 5 × 5-μm openings (channels) between them using polydimethylsiloxane. The design and fabrication of this microchannel 2-dimensional (2D) filter device and configuration of the experimental setup have been described previously in detail.16
To establish the flow of sample through the 2D filter device, the outlet of the device was connected to a waste reservoir (60-mL syringe barrel; Becton Dickinson) with a 60-cm long piece of PE-60 tubing (Instech Laboratories) filled with HBSS with Ca++ and Mg++. The difference between the level of liquid in the inlet reservoir of the 2D filter device and the level of liquid in the waste reservoir provided the driving pressure. The zero pressure difference corresponded to the absence of movement of RBCs within the device. RBCs (8 μL, 20% hematocrit) were loaded into the inlet reservoir and allowed to enter the network (approximately 1 minute) by lowering the waste reservoir tubing to a position located approximately 15 cm below the zero pressure point. Once RBCs entered the capillary area, the waste reservoir tubing was raised to approximately 38 mm below the zero pressure point, which allowed RBCs to pass through the 25 μm length of the capillary in approximately 2-3 seconds. The height of the reservoir was then left unchanged throughout the experiment. The passage of RBCs through the 2D filter was recorded using a 40×/0.75 (NA) Ph2 Plan Fluorite objective on a TE300 Nikon inverted microscope, using a Retiga Exi (QImaging) camera controlled by iVision 4.01 (BioVision) at a rate of 10 frames/s. The images were analyzed frame by frame to measure the time from RBC channel contact to egress, and the results expressed as seconds per passage. Differences in passage time between the control and CR1-ligated RBCs with and without inhibitors were compared using the Mann-Whitney test (Prism 4.0). A P value ≤ .05 was considered statistically significant.
Flow cytometry
RBCs were fixed and permeabilized as described in,7 and incubated for 15 minutes with Ab (as noted in the figure legends) in 0.5% BSA in HBSS buffer at 4°C, followed by 2 washes and incubation for 15 minutes with AlexaFluor488–labeled secondary Ab specific for each primary Ab at a dilution recommended by the manufacturer. In each experiment, at least 10 000 events were recorded using FACScan or LRSII (Becton Dickinson) and analyzed using CellQuest Pro Version 4.0.1 software (BD Biosciences).
Statistical analysis
The statistical analysis tests were performed using Prism Version 4.0 (GraphPad Software) and are detailed for each experiment.
Results
Ligation of RBC CR1 induces Ca++ influx
RBCs are classically described as passive carriers of immune complexes from blood to resident macrophages in the liver and spleen. We have previously reported that the ligation of RBC CR1 by complement-opsonized particles reorganizes CR1 into large clusters.7 Here, we asked whether ligation of CR1 would trigger a RBC Ca++ influx. Unlike any other CR1-bearing cell type, the expression patterns of CR1 on RBCs are genetically determined with RBCs expressing approximately 100 CR1/cell for low CR1 expressors, 500/cell on intermediate CR1 expressors, and 1000/cell for high CR1 expressors.17 To determine whether the levels of CR1 expression on RBCs correlate with the magnitude of CR1-mediated Ca++ influx, we used RBCs from donors with known low and high RBC CR1 expression.7 RBCs were loaded with Fluo-4 and incubated with either irrelevant match control Ab, anti-CD47, or anti-CR1 mAb, and the results analyzed by flow cytometry. Our results show that ligation of RBC CR1 with mAb (Figure 1A) promoted a sustained Ca++ influx, with RBCs from high CR1 donors displaying a more pronounced Ca++ influx (300nM) compared with RBCs from low CR1 expressors (75nM). Ligation of RBCs from either donor with anti-CD47 mAb did not trigger any measurable Ca++ influx (data not shown). When experiments were repeated using complement-opsonized beads as physiologic CR1 ligands (Figure 1B), a similar RBC Ca++ influx pattern was observed, although the amplitude and the differences between RBC Ca++ influx of high (100nM) and low CR1 expressors (60nM) were less pronounced. Similarly, ligation of RBC CR1 with Abs rendered a higher ratio between CR1 levels of high CR1 donors (mean fluorescence intensity [MFI] = 108) and low CR1 donors (MFI = 7.23, control Ab MFI = 2.23) (Figure 1C) compared with binding of the fluorescently labeled complement opsonized beads to high CR1 RBCs (MFI = 440; Figure 1D) and low CR1 RBCs (MFI = 110, control beads MFI = 3.55; Figure 1D). Our results suggest that, although the RBC CR1 levels vary by approximately 10-fold between high and low CR1 expressors, from a functional perspective the gap is less pronounced (Figures 1E-F). Because carrying immune-complexes and complement-opsonized particles such as viruses and bacteria could augment the Ca++ influx promoted by shear stress experienced by circulating RBCs,18,19 we next searched for an RBC Ca++ channel that is also a mechanoreceptor.

Ligation of RBC CR1 promotes Ca++ influx that depends on the genetically determined CR1 levels. (A) Antibody ligation of RBC CR1 triggers a Ca++ influx. Intra-RBC Ca++ concentration of Fluo-4–loaded RBCs from known high and low CR1 expressors was measured by flow cytometric analysis for 20 seconds before RBC CR1 was ligated (arrow) by anti-CR1 mAb. Changes in intra-RBC Ca++ concentration were measured for a total of 2 minutes. (B) Ligation of CR1 by complement opsonized-particles promotes RBC Ca influx. Intra-RBC Ca++ concentration of Fluo-4–loaded RBCs from known high and low CR1 expressors was measured by flow cytometry for 20 seconds before RBC CR1 was ligated (arrow) by control or complement opsonized beads. Each line represents the average RBC fluorescence analyzed at a rate of approximately 1000 RBC/second. (C) Characterization of RBC CR1 levels by antibody shows large gap between low and high CR1 expressors. RBCs from known low and high CR1 expressors were incubated with anti-CR1 mAb followed by AlexaFluor488 secondary Ab and analyzed by flow cytometry. (D) Functional characterization of RBC CR1 using complement-opsonized beads identifies a narrower gap between RBCs from high and low expressors. RBCs from known low and high CR1 expressors were incubated with fluorescently labeled complement opsonized beads for 30 minutes, washed, and analyzed by flow cytometry. (E) Fluorescence microscopy overlapped with phase contrast microscopy of RBCs from a high CR1 expressor after incubation with fluorescently labeled complement opsonized beads. (F) Fluorescence microscopy overlapped with phase contrast microscopy of RBCs from a low CR1 expressor after incubation with fluorescently labeled complement opsonized beads. Bar represents 10 μm. The results shown here are from the same individuals and are representative of 3 independent experiments using 3 different high CR1 and 2 different low CR1 expressors.
Ligation of RBC CR1 promotes Ca++ influx that depends on the genetically determined CR1 levels. (A) Antibody ligation of RBC CR1 triggers a Ca++ influx. Intra-RBC Ca++ concentration of Fluo-4–loaded RBCs from known high and low CR1 expressors was measured by flow cytometric analysis for 20 seconds before RBC CR1 was ligated (arrow) by anti-CR1 mAb. Changes in intra-RBC Ca++ concentration were measured for a total of 2 minutes. (B) Ligation of CR1 by complement opsonized-particles promotes RBC Ca influx. Intra-RBC Ca++ concentration of Fluo-4–loaded RBCs from known high and low CR1 expressors was measured by flow cytometry for 20 seconds before RBC CR1 was ligated (arrow) by control or complement opsonized beads. Each line represents the average RBC fluorescence analyzed at a rate of approximately 1000 RBC/second. (C) Characterization of RBC CR1 levels by antibody shows large gap between low and high CR1 expressors. RBCs from known low and high CR1 expressors were incubated with anti-CR1 mAb followed by AlexaFluor488 secondary Ab and analyzed by flow cytometry. (D) Functional characterization of RBC CR1 using complement-opsonized beads identifies a narrower gap between RBCs from high and low expressors. RBCs from known low and high CR1 expressors were incubated with fluorescently labeled complement opsonized beads for 30 minutes, washed, and analyzed by flow cytometry. (E) Fluorescence microscopy overlapped with phase contrast microscopy of RBCs from a high CR1 expressor after incubation with fluorescently labeled complement opsonized beads. (F) Fluorescence microscopy overlapped with phase contrast microscopy of RBCs from a low CR1 expressor after incubation with fluorescently labeled complement opsonized beads. Bar represents 10 μm. The results shown here are from the same individuals and are representative of 3 independent experiments using 3 different high CR1 and 2 different low CR1 expressors.
TRPC1 is involved in the Ca++ influx induced by CR1 ligation and shear stress
To verify the putative involvement of a stretch-activated cation channel (SAC) in CR1-mediated Ca++ influx, we first used GsMTx-4, a nonspecific peptide blocker of cationic SAC.20 Our data showed that preincubation of RBCs with increasing concentrations of GsMTx-4 (1μM and 5μM) progressively inhibited CR1 mediated Ca++ influx as measured by flow cytometry (Figure 2A). We next focused on the widely expressed stretch-activated cation channel, TRPC1, which was shown to promote Ca++ influx after mechanical stimulation of cell membranes.21,22 We started by investigating the presence of TRPC1 in RBCs by using immunofluorescence microscopy, flow cytometry, and immunoblotting methods. Fixed RBCs were permeabilized and reacted with either rabbit control or rabbit anti-TRPC1 mAb, followed by goat anti-rabbit AlexaFluor488 conjugated IgG. Immunofluorescence microscopy demonstrated a distinct punctate pattern at the plasma membrane, uniformly distributed throughout the RBC population (Figure 2B). To further confirm the presence of TRPC1 in RBCs, we used immunoblot analysis of RBC lysates probed with rabbit anti-TRPC1 mAb (Santa Cruz Biotechnology). Our results show that anti-TRPC1 pAb detected a protein running at a MW around 85 kDa (Figure 2C right lane), which corresponded to the MW of TRPC1 identified in testes tissue lysate (Santa Cruz Biotechnology) used as a positive control (Figure 2C left lane).23 In addition, analysis of RBCs from 2 low and 3 high CR1 donors by flow cytometry showed no consistent differences in RBC TRPC1 expression levels, further suggesting that the differences measured in Ca++ influx between various RBC donors are due to different expression patterns of CR1 and not TRPC1 (supplemental data, available on the Blood Web site; see the Supplemental Materials link at the top of the online article; and Figure 2A).

RBCs express functional TRPC1. (A) RBC CR1 ligation promotes a SAC-dependent Ca++ influx. RBCs were loaded with Fluo-4 and incubated with buffer, 1 or 5μM GsMTX-4 for 30 minutes and then analyzed by flow cytometry for CR1-mediated Ca++ influx (arrow) as described above. (B) RBCs express TRPC1. RBCs were fixed, permeabilized, and incubated with either control (left panel) or rabbit monoclonal anti-TRPC1 (right panel) for 16 hours, washed, and incubated with goat anti–rabbit AlexaFluor488. Cells were imaged under the microscope using the fluorescein isothiocyanate/green fluorescent protein filter. (C) Immunoblotting detection of TRPC1 in RBCs. Testes tissue lysate (Santa Cruz Biotechnology; second lane) and RBC lysate (third lane) were separated by electrophoresis using Tris-HCl gels and probed with rabbit anti-TRPC1 Ab (Santa Cruz Biotechnology). TRPC1 is seen as a band at approximately 87 kDa (arrow) in both positive control and RBC lysate. Lane 1, Bio-Rad molecular weight markers. (D) Anti-TRPC1 inhibitory antibody T1E3 binds RBCs. RBC were incubated with either 1/500 dilution of normal rabbit serum (dotted histogram) or T1E3 Ab anti-serum (continuous histogram) for 10 minutes followed by AlexaFluor488 goat anti–rabbit. Cells were washed and examined by flow cytometry. (E) CR1 ligation triggers a Ca++ influx dependent on TRPC1. Fluo-4 loaded RBCs were preincubated with control serum or T1E3 for 20 minutes, washed twice, and then analyzed by flow cytometry for CR1-mediated Ca++ influx (arrow) as described above.
RBCs express functional TRPC1. (A) RBC CR1 ligation promotes a SAC-dependent Ca++ influx. RBCs were loaded with Fluo-4 and incubated with buffer, 1 or 5μM GsMTX-4 for 30 minutes and then analyzed by flow cytometry for CR1-mediated Ca++ influx (arrow) as described above. (B) RBCs express TRPC1. RBCs were fixed, permeabilized, and incubated with either control (left panel) or rabbit monoclonal anti-TRPC1 (right panel) for 16 hours, washed, and incubated with goat anti–rabbit AlexaFluor488. Cells were imaged under the microscope using the fluorescein isothiocyanate/green fluorescent protein filter. (C) Immunoblotting detection of TRPC1 in RBCs. Testes tissue lysate (Santa Cruz Biotechnology; second lane) and RBC lysate (third lane) were separated by electrophoresis using Tris-HCl gels and probed with rabbit anti-TRPC1 Ab (Santa Cruz Biotechnology). TRPC1 is seen as a band at approximately 87 kDa (arrow) in both positive control and RBC lysate. Lane 1, Bio-Rad molecular weight markers. (D) Anti-TRPC1 inhibitory antibody T1E3 binds RBCs. RBC were incubated with either 1/500 dilution of normal rabbit serum (dotted histogram) or T1E3 Ab anti-serum (continuous histogram) for 10 minutes followed by AlexaFluor488 goat anti–rabbit. Cells were washed and examined by flow cytometry. (E) CR1 ligation triggers a Ca++ influx dependent on TRPC1. Fluo-4 loaded RBCs were preincubated with control serum or T1E3 for 20 minutes, washed twice, and then analyzed by flow cytometry for CR1-mediated Ca++ influx (arrow) as described above.
To investigate the functional involvement of TRPC1 in CR1-mediated Ca++ influx, we next used a rabbit anti-sera (T1E3) that was raised against the extracellular portion of TRPC1 and was used successfully to inhibit TRPC1-dependent Ca++ influx.24 Our results showed that T1E3 recognized an epitope on RBCs (Figure 2D) and that preincubation of RBCs with T1E3 before CR1 ligation successfully prevented CR1-mediated Ca++ influx (Figure 2E). Importantly, sera had to be removed before CR1 ligation by repeated washes to prevent RBC Ca++ influx followed by RBC lysis due to activation of the classical complement pathway and formation of C5b-9 complex in RBC plasma membrane. Taken together, our results strongly suggest that TRPC1 is implicated in the CR1-mediated RBC Ca++ influx.
CR1 ligation increases RBC membrane deformability
The CR1-mediated immune clearance process takes place in liver and spleen where RBCs are forced to pass through narrow 3- to 4-μm capillaries as well as small 2- to 3-μm slits in the walls of the reticuloendothelial sinusoids.25 Therefore, we hypothesized that CR1 ligation would increase RBC membrane deformability thus offering mechanical advantages during the immune complex clearing process. To address this question, we used 2 methods for measuring membrane deformability: (1) shape recovery after LOT-assisted cell deformation and (2) filtration through microchannel arrays. In the first approach, the deformability of RBCs is measured as the time it takes the cell to recover its original biconcave shape after a bead attached to its membrane by anti-glycophorin C mAb is pulled away to deform the cell.26-29 The second approach uses microchannel arrays to assess RBC deformability based on the time required for RBCs to squeeze in and pass through an array of capillary-like channels.30 This method requires no RBC manipulation and can analyze large numbers of RBCs within minutes after blood collection. Using LOT, the force necessary to elongate the RBCs by 2 μm was 9-13 pN and showed no significant difference between control and CR1-cross-linked RBCs, independent of the presence of extracellular Ca++ (supplemental Figure 3A). A range of force values was observed, since the elongation was held constant, and the variation in force was coupled to the variation in the cell size; our observed force of approximately 10pN is similar to other reports31,32 and demonstrates that small extensions were used. We found that ligation of CR1 with primary and cross-linking secondary Ab shortened significantly the recovery time (60 ± 10 milliseconds, P < .01; Figure 3A) compared with the control cells without CR1 cross-linking (72 ± 11 milliseconds; Figure 3A). The decreased recovery time induced by CR1 cross-linking did not occur when extracellular Ca++ was chelated in Mg++-EGTA (ethyleneglycoltetraacetic acid) buffer (74 ± 10 milliseconds, P < .01; Figure 3A). Shape recovery after extension measures the dynamic response of the cell. Thus, the extensional recovery time depends on RBC extensional rigidity and on viscous dissipation in the membrane and the cytoplasm. Because the forces required to extend both CR1-ligated and control RBCs were the same, our results indicate that the faster recovery time promoted by CR1 ligation is likely the result of decreased cell viscosity and not increased extensional elasticity.

Ligation of CR1 increases RBC membrane deformability and depends on extracellular Ca++. (A) CR1-mediated increase in membrane deformability depends on Ca++. Freshly isolated RBCs were incubated with either control Ab or anti-CR1 mAb in the presence of Ca++ and Mg++ (Ca++) or Mg++-EGTA (EGTA) for 10 minutes at RT. RBCs were washed, and 30 cells were analyzed for each condition by LOT. The recoil time (tc) of CR1-ligated RBCs (60 ± 2 milliseconds) was significantly shorter (P < .001) than the recoil time of control RBCs (72 ± 3 milliseconds). The bars represent 95% confidence interval. (B) Ligation of RBC CR1 decreases the time required for RBC to pass through microchannels. Sequential images (3 frames/s) of a RBC entering the microfluidic device are shown in the top panel. The extent of the decrease in the time required for RBCs to pass through the microchannels depends directly on the number of CR1 molecules on the surface of RBCs. Each dot in the graph represents a RBC. These results are representative of 5 independent experiments using different anti-CR1 Abs and different high and low CR1 donors.
Ligation of CR1 increases RBC membrane deformability and depends on extracellular Ca++. (A) CR1-mediated increase in membrane deformability depends on Ca++. Freshly isolated RBCs were incubated with either control Ab or anti-CR1 mAb in the presence of Ca++ and Mg++ (Ca++) or Mg++-EGTA (EGTA) for 10 minutes at RT. RBCs were washed, and 30 cells were analyzed for each condition by LOT. The recoil time (tc) of CR1-ligated RBCs (60 ± 2 milliseconds) was significantly shorter (P < .001) than the recoil time of control RBCs (72 ± 3 milliseconds). The bars represent 95% confidence interval. (B) Ligation of RBC CR1 decreases the time required for RBC to pass through microchannels. Sequential images (3 frames/s) of a RBC entering the microfluidic device are shown in the top panel. The extent of the decrease in the time required for RBCs to pass through the microchannels depends directly on the number of CR1 molecules on the surface of RBCs. Each dot in the graph represents a RBC. These results are representative of 5 independent experiments using different anti-CR1 Abs and different high and low CR1 donors.
Next, we used RBCs from donors with known low and high RBC CR1 expression7 to determine whether the levels of CR1 expression on RBCs correlate with the magnitude of CR1-mediated increases in RBC membrane deformability. RBCs from low and high CR1 expressors were ligated with primary anti-CR1 Ab and cross-linking secondary Ab at 4°C, warmed to 37°C for 10 minutes, and then the time required for RBCs to fold, pass through and completely exit the 25-μm channels was measured and expressed as one value per RBC (Figure 3B top panel). Our results (Figure 3B bottom panel) show that the number of CR1 molecules per RBC positively correlated with the increase in membrane deformability. High CR1 expressing RBCs required approximately half the time (t = 0.78 ± 0.14 seconds) to pass through microchannels compared with low CR1 expressing RBCs (t = 1.42 ± 0.22 seconds) and approximately a third of the time required for the control RBCs (t = 2.21 ± 0.18 seconds). There were no statistical differences between the transit times of RBCs from high and low CR1 donors treated with control Ab (data not shown). In addition, all 3 steps (folding, passage, and exit) were equally affected by ligation of CR1. One possible alternative explanation for the shortened transit time of CR1-ligated RBCs could be that the volume of RBCs significantly decreased after CR1 ligation through a Ca++-dependent K+-modulated mechanism (Gardos effect).33 Therefore, we also quantified the relative size of RBCs using flow cytometry and forward light scattering detection during CR1 ligation. Our results (supplemental Figure 3B) show that CR1 ligation did not significantly affect the size of RBCs, further suggesting that CR1 ligation affected RBC deformability directly.
RBC CR1 ligation induces phosphorylation of β-spectrin and α-adducin
The RBC membrane skeleton comprises spectrin tetramers arranged to form an elastic network (reviewed in Mohandas and Gallagher34 ). Serine phosphorylation of β-spectrin is one known means of modulating the mechanical properties of the RBC membrane.9 Based on our findings regarding the effect of CR1 ligation by Ab on RBC membrane deformability (Figure 3), we hypothesized that ligation of CR1 would promote phosphorylation of β-spectrin. To assess this hypothesis, CR1 was ligated with primary and secondary Ab at 4°C, and phosphorylation levels of spectrin were measured by both fluorescence-activated cell sorting (FACS) and Western blotting after incubation of RBCs at 37°C for 0, 10, and 25 minutes. These time points were chosen knowing that more than 90% of the immune complexes are removed in the first 10 minutes after appearing in the circulation.35,36 For FACS analysis, CR1 ligation was performed using rabbit polyclonal Ab followed by incubation with highly cross-absorbed goat anti–rabbit Ab that lacks cross-reactivity with anti-phospho-serine/-threonine detection Abs. After incubation with anti-CR1 Ab or control IgG (Figure 4A), RBCs were fixed and permeabilized, and total phosphorylation levels were detected by anti-phosphoserine/-threonine mAb, followed by AlexaFluor488 goat anti–mouse IgG. Our results (Figure 4A) show that total phosphorylation levels of serine/threonine residues after CR1 ligation were elevated after 10 minutes and decreased to almost basal values in 25 minutes. We next used Western blot analysis to specifically evaluate the levels of phospho β-spectrin in RBC lysates obtained at 0, 10, and 25 minutes after CR1 ligation. Consistent with the FACS results, the phosphorylation levels of β-spectrin reached a maximum at 10 minutes and decreased to or below normal values after 25 minutes (Figure 4A bottom panel). In parallel, we investigated the effect of RBC CR1 levels on the phosphorylation levels of β-spectrin using known CR1 expressors. We found that at 10 minutes the phosphorylation levels of β-spectrin were higher in high CR1 expressors (supplemental Figure 4), although at earlier and later time points the differences were not significant (data not shown).

Ligation of RBC CR1 induces serine/threonine phosphorylation of β-spectrin and adducin. (A) Cross-linking of RBC CR1 induces time-dependent serine/threonine phosphorylation, as assessed by FACS. RBCs were incubated with either control Ab or anti-CR1 for various amounts of time, fixed, permeabilized, and serine/threonine phosphorylation levels assessed by flow cytometry. (B) Western blotting analysis of the serine/threonine phosphorylation of β-spectrin (arrow) was temporally concordant with the FACS analysis. Control represents the basal serine/threonine phosphorylation levels of β-spectrin in fresh RBCs at 4°C. The experiment was repeated 4 times with similar results. (C) Phosphorylation levels of adducin increases 10 minutes after ligation of CR1. Control represents basal levels of phospho-adducin (lane 1), and as positive control, RBCs were activated by 0.5μM PMA (lane 3). Extra bands present in the anti-CR1 lane represent heavy and light IgG chains or the cross-linking anti-CR1 Ab. Loading controls are represented by the total amount of adducin in RBC lysate probed with rabbit polyclonal antibody anti-adducin. These results are representative of 3 independent experiments. (D) CR1-mediated adducin phosphorylation depends on Ca++. RBCs were incubated with PMA or anti-CR1 in the presence or absence of Ca++ (Mg++ EGTA) and phosphorylation levels of adducin measured as above. Loading controls are represented by the levels of glyceraldehyde-3-phosphate dehydrogenase in RBC lysates.
Ligation of RBC CR1 induces serine/threonine phosphorylation of β-spectrin and adducin. (A) Cross-linking of RBC CR1 induces time-dependent serine/threonine phosphorylation, as assessed by FACS. RBCs were incubated with either control Ab or anti-CR1 for various amounts of time, fixed, permeabilized, and serine/threonine phosphorylation levels assessed by flow cytometry. (B) Western blotting analysis of the serine/threonine phosphorylation of β-spectrin (arrow) was temporally concordant with the FACS analysis. Control represents the basal serine/threonine phosphorylation levels of β-spectrin in fresh RBCs at 4°C. The experiment was repeated 4 times with similar results. (C) Phosphorylation levels of adducin increases 10 minutes after ligation of CR1. Control represents basal levels of phospho-adducin (lane 1), and as positive control, RBCs were activated by 0.5μM PMA (lane 3). Extra bands present in the anti-CR1 lane represent heavy and light IgG chains or the cross-linking anti-CR1 Ab. Loading controls are represented by the total amount of adducin in RBC lysate probed with rabbit polyclonal antibody anti-adducin. These results are representative of 3 independent experiments. (D) CR1-mediated adducin phosphorylation depends on Ca++. RBCs were incubated with PMA or anti-CR1 in the presence or absence of Ca++ (Mg++ EGTA) and phosphorylation levels of adducin measured as above. Loading controls are represented by the levels of glyceraldehyde-3-phosphate dehydrogenase in RBC lysates.
In human RBCs, PMA-induced protein kinase C-mediated phosphorylation of α-adducin at Ser 726 promotes dissociation of actin from the spectrin network,11 and it contributes to increasing RBC membrane deformability.37 Therefore, we next analyzed the phosphorylation status of α-adducin after RBC CR1 ligation using a specific mAb that recognized the phosphorylation of human adducin at Ser 726 (Figure 4C). Total adducin in RBC lysates was detected on a parallel gel using an anti-adducin mAb (Figure 4C bottom panel). The basal level of phospho-Ser 726 α-adducin is usually undetectable without the use of calyculin, which blocks phosphatase activity.11 We did not use calyculin for these experiments, as it is likely that ligation of CR1 triggers a cascade of both phosphorylation and dephosphorylation events and blocking just one arm of this process could bias the detection of the actual signal. Our data show that ligation of CR1 induced a significant phosphorylation of adducin at Ser 726 at 10 minutes (Figure 4B). The finding that the CR1-mediated increase in membrane deformability requires Ca++ prompted us to investigate the effect of lack of Ca++ on CR1-mediated phosphorylation of adducin at Ser 726. Western blot analysis showed (Figure 4D) that unlike PMA-induced phosphorylation of adducin, which is independent of Ca++, removal of extracellular Ca++ partially inhibited CR1-mediated adducin phosphorylation. These phosphorylation events could explain, along with phosphorylation of β-spectrin, the increase in RBC membrane deformability measured by LOT and microchannels and the Ca++ requirements. The earlier (5 minutes) and later (25 minutes) time points did not show any detectable phosphorylation of adducin at Ser 726 (data not shown). The effect of RBC CR1 levels on phosphorylation of adducin at Ser 726 was much less pronounced than the one observed on β-spectrin (supplemental Figure 4B), suggesting a lesser contribution of phosphorylated adducin to total RBC deformability as assessed by our 2 methods.
Inhibition of casein kinase 2 prevents CR1-mediated increase in RBC membrane deformability
Because β-spectrin contains specific phosphorylation sites for both CKI (casein kinase I) and CKII (casein kinase II),38 we investigated the role of each of these enzymes in CR1-mediated spectrin phosphorylation using specific inhibitors. RBCs were incubated for 15 minutes before CR1 ligation with either 0.2% dimethyl sulfoxide, D4476, a CKI inhibitor,39 or DMAT, a CKII inhibitor.40 The effects of CK inhibitors on CR1-initiated β-spectrin phosphorylation were assessed by Western blotting using anti-phosphoserine/threonine Ab. The CR1-mediated increase in β-spectrin phosphorylation was sensitive to CKII inhibition (Figure 5A lane 4), but not to CKI inhibition (Figure 5A lane 3). Unexpectedly, when CR1 was ligated in the presence of the CKI inhibitor, phosphorylation was above basal levels (Figure 5A lane 3). To further confirm our findings, we performed deformability studies on CR1-ligated RBCs treated with the CKI or CKII inhibitors using filtration through microchannel arrays. Consistent with our Western blot data, we found that CKII inhibitors effectively prevented the CR1-mediated increase in membrane deformability (P < .001), whereas CKI inhibitors had no measurable effect (P = .76; Figure 5B).

Ligation of CR1 promotes β-spectrin serine phosphorylation through a CK II-dependent mechanism. (A) RBCs were incubated with anti-CR1 mAb in the presence or absence of CK I or CK II inhibitors for 15 minutes at 4°C. RBCs were then washed and cross-linked for an additional 15 minutes with goat anti-mouse secondary Ab at 37°C. RBCs were washed, lysed, and tested for serine phosphorylation by immunoblotting with an anti-phospho-serine/-threonine Ab. Bottom panel shows β-actin as loading control. The experiments were done twice with similar results. (B) Inhibition of CK II blocks the effect of CR1 ligation on RBC membrane deformability. RBCs were incubated with anti-CR1 mAb followed by secondary cross-linking Ab in the presence of CK I or CKII inhibitors, and deformability was tested using microfluidic devices within 15 minutes. These results are representative of 2 independent experiments
Ligation of CR1 promotes β-spectrin serine phosphorylation through a CK II-dependent mechanism. (A) RBCs were incubated with anti-CR1 mAb in the presence or absence of CK I or CK II inhibitors for 15 minutes at 4°C. RBCs were then washed and cross-linked for an additional 15 minutes with goat anti-mouse secondary Ab at 37°C. RBCs were washed, lysed, and tested for serine phosphorylation by immunoblotting with an anti-phospho-serine/-threonine Ab. Bottom panel shows β-actin as loading control. The experiments were done twice with similar results. (B) Inhibition of CK II blocks the effect of CR1 ligation on RBC membrane deformability. RBCs were incubated with anti-CR1 mAb followed by secondary cross-linking Ab in the presence of CK I or CKII inhibitors, and deformability was tested using microfluidic devices within 15 minutes. These results are representative of 2 independent experiments
Discussion
The data presented in this study identify a novel functional consequence of RBC CR1 engagement that is likely relevant to the immune complexes transfer process. We show that CR1 ligation increases RBC membrane deformability by promoting β-spectrin phosphorylation through activation of CKII. The present work is based on our previous findings that showed that CR1 on circulating RBC clusters only upon ligation of complement-opsonized particles.7 To investigate further the functional consequences of RBC CR1 ligation, we initially assessed the phosphorylation status of CR1 cytoplasmic tail after CR1 ligation. Although CR1 has a CKII phosphorylation site in its cytoplasmic domain, we and others were unable to document changes in RBC CR1 phosphorylation status after either exposure to PMA or CR1 ligation41 (unpublished results).
The most important function of RBCs, delivering oxygen to tissues, depends largely on the deformability of their membranes, which is a measure of the ability of RBCs to change shape in response to external forces.42,43 The dynamic, energy-dependent link between cell membrane and skeleton is essential for the ability of RBCs to pass rapidly through capillaries.44,45 Maintaining optimal RBC membrane deformability is thus critical for the body to insure appropriate perfusion of the tissues by preventing trapping of less deformable RBCs in capillaries. Therefore, we next investigated the effect of CR1 ligation on RBC membrane deformability by using 2 complementary methods: (1) LOT shape recovery and (2) filtration through microchannel networks. Shape recovery and filterability measure 2 essential elastic properties of the cell, dynamic extensional elasticity, and bending elasticity. Our hypothesis is that engagement of RBC CR1 by complement-opsonized particles would increase membrane deformability to facilitate the delivery of opsonized particles to resident macrophages present in the narrow (3-4 μm) sinusoid capillaries in the liver and spleen.25 Our results show that ligation of CR1 significantly decreases RBC extensional recovery time and RBC passage time through microchannels, which importantly, also depends on the number of RBC CR1 molecules (Figure 3). However, we believe that it is unlikely that RBCs expressing a genetically determined high number of CR1 molecules would have a significant advantage over RBCs expressing a low number of CR1 in normal conditions.
Previous studies demonstrated that increased RBC membrane tension by mechanical stimulation was responsible for augmented membrane permeability for Ca++.46 Here, we addressed the involvement of stretch-activated ion channel TRPC1 in CR1-mediated Ca++ influx, a protein that has been proposed to be involved in transducing membrane stretch into Ca++ influx.22 Our results obtained using an inhibitory antibody against TRPC1 (T1E3) show for the first time that TRPC1 is functionally expressed in mature RBCs and is involved in mediating CR1-dependent Ca++ influx. Our data also show that the levels of TRPC1 expression in RBCs from low and high CR1 expressors did not follow the expression pattern of CR1. This finding strongly suggests that the amplitude of the RBC Ca++ influx promoted either by anti-CR1 mAb (Figure 1A) or complement-opsonized particles (Figure 1B) is due to the actual number of CR1 molecules per RBC and not to the expression patterns of the TRPC1.
We and others have shown that engagement of RBC GPA (glycophorin A) by C3 complement fragments during complement activation significantly reduces RBC membrane deformability, which likely accelerates RBC removal from the circulation.26,47 As the signaling events triggered by complement fragments engaging CR1 and GPA have opposing consequences on RBC membrane deformability, the ratio between the number of complement-ligated CR1 and GPA molecules could be important in determining the net change in RBC membrane deformability. Consequently, the genetically determined number of CR1 molecules RBCs could be relevant in certain pathologic conditions associated with significant intravascular complement activation, such as sepsis (reviewed in Markiewski et al48 ) with high RBC CR1 expressors having functional advantage over the low RBC CR1 expressors. Consistent with our hypothesis, a recent clinical study extended over 5 years compared the mortality and morbidity of chronically dialyzed patients with consistently low or high RBC CR1 levels. Patients with consistently low RBC CR1 levels had a 1.6× higher mortality compared with those with high RBC CR1 levels, likely due to diminished immune-clearance.49
The role of adducin phosphorylation by PKC in modulating RBC membrane deformability and stability is well described in the literature.11,37 We showed here that CR1 ligation leads to increased membrane deformability and increased phosphorylation of α-adducin and β-spectrin (Figures 3–4). Interestingly, RBCs from the Rac1−/−; Rac2−/− mouse, showing increased basal phosphorylation of α-adducin at the site comparable with human Ser-726, have significantly decreased membrane deformability.44 There could be 2 reasons for this apparent discrepancy between normal human and mouse Rac1−/−; Rac2−/− RBCs. First, the Rac1−/−; Rac2−/− mouse may have an abnormally high actin-to-spectrin ratio that could directly cause cytoskeletal rearrangement; and second, the microcytic anemia observed in the Rac1−/−; Rac2−/− mouse may cause an apparent decrease in membrane deformability. Importantly, the effect of CR1 ligation on membrane deformability lasted significantly longer than its effect on spectrin phosphorylation (Figure 4), which would suggest that additional regulatory mechanisms such as interaction of spectrin with certain lipids from plasma membrane are also important in modulating RBC membrane deformability and stability.50,51
Our studies using cell-permeant specific inhibitors for casein kinases identified CKII as the main kinase responsible for phosphorylation of β-spectrin promoted by CR1 ligation and detected no effect due to CKI inactivation (Figure 5). However, there are conflicting reports in the literature regarding the identity of the RBC CK responsible for phosphorylation of β-spectrin, with one report identifying CK II38 and another one CKI.9 This seeming contradiction may be explained by the fact in the paper identifying CKI as the main β-spectrin kinase, the authors used: (1) [γ32P] ATP as a phosphoryl donor, which can be used by both CKI and CKII and (2) N-(2-aminoethyl)-5-chloroisoquinoline-sulfonamide, an inhibitor now known to inhibit ribosomal S6 kinase, serum/glucocorticoid regulated kinase, and stress/mitogen-activated kinase, in addition to CKI.39,52 Intriguingly, our data showed that incubation of RBCs with CKI inhibitor increased the levels of β-spectrin phosphorylation, both in control and CR1-ligated RBCs (Figure 5). This unexpected result could be explained by the regulatory activity of CKI on serine/threonine protein phosphatases present in RBCs and responsible for dephosphorylation of spectrin.53,54
In conclusion, we have demonstrated that ligation of RBC CR1 triggers a complex Ca++-dependent signaling cascade leading to a significant increase in plasma membrane deformability. In addition, recognition of CR1 ligation, as well as its expression levels as important factors involved in regulating RBC membrane deformability will allow us a better understanding of immune adhesion as well as the immune transfer process. Based on our work, one can also speculate that in critically ill patients transfusion with RBCs from high CR1 expressors could be potentially advantageous especially in pathologies associated with significant complement activation.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
We thank Henry Marsh for providing the anti-CR1 mAb, Xiaoqiang Yao for providing the T1E3 anti-TRPC1 pAb, and Peter Weller for critically reviewing the manuscript.
This work was supported by National Institutes of Health grants AI42987 (A.N.W) and R01HL32854 (D.E.G). Part of the work was supported by the W81XWH 07-1-0286 grant from the Medical Research and Material Command (MRMC) and FA9550 08-1-0364 grant from the Defense Advanced Research Projects Agency.
National Institutes of Health
Authorship
Contribution: I.C.G. designed the research, performed the majority of the experiments, analyzed the data, and wrote the manuscript; A.M.G., R.M., S.S.S., and J.A.K. designed the research and performed experiments; A.N.-W. and D.E.G. analyzed data and critically reviewed the manuscript; and J.B. provided technical assistance for red cell deformability experiments.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Ionita C. Ghiran, Division of Allergy & Inflammation, Beth Israel Deaconess Medical Center, Harvard Medical School, Center for Life Science, CLS 930, 3 Blackfan Cir, Boston, MA 02115; e-mail: ighiran@bidmc.harvard.edu.
