

Abstract
Bone marrow mesenchymal cells (MSCs) can protect leukemic cells from chemotherapy, thus increasing their survival rate. We studied the potential molecular mechanisms underlying this effect in acute lymphoblastic leukemia (ALL) cells. Coculture of ALL cells with MSCs induced on the lymphoblast plasma membrane the expression of a signaling complex formed by hERG1 (human ether-à-go-go-related gene 1) channels, the β1-integrin subunit, and the chemokine receptor CXC chemokine receptor-4. The assembly of such a protein complex activated both the extracellular signal-related kinase 1/2 (ERK1/2) and the phosphoinositide 3-kinase (PI3K)/Akt prosurvival signaling pathways. At the same time, ALL cells became markedly resistant to chemotherapy-induced apoptosis. hERG1 channel function appeared to be important for both the initiation of prosurvival signals and the development of drug resistance, because specific channel blockers decreased the protective effect of MSCs. NOD/SCID mice engrafted with ALL cells and treated with channel blockers showed reduced leukemic infiltration and had higher survival rates. Moreover, hERG1 blockade enhanced the therapeutic effect produced by corticosteroids. Our findings provide a rationale for clinical testing of hERG1 blockers in the context of antileukemic therapy for patients with ALL.
Introduction
Cure rates for children with acute lymphoblastic leukemia (ALL), the most common pediatric malignancy, have improved markedly over the past 2 decades, approaching 90% in some trials.1 Despite this success, a subset of patients remain refractory to chemotherapy,2,3 while others require prolonged and intensive treatment to achieve continuous remission, which may lead to serious sequelae.4
It has been reported that bone marrow mesenchymal cells (MSCs) can contribute to drug resistance in leukemic cells.5-11 Several mechanisms have been proposed to explain this effect, such as the production of asparagine by MSCs (which protects ALL cells from asparagine depletion by asparaginase),12 and the interaction between the vascular cell adhesion molecule-1 (VCAM-1) on MSCs and the very late antigen-4 (VLA-4) integrin on leukemic cells resulting in protection against cytarabine and etoposide cytotoxicity.13 Other molecular interactions might also play a role, including the interplay between stromal-derived factor 1α (SDF-1α) and its receptor, CXC chemokine receptor-4 (CXCR4).14 Both integrin and CXCR4 signaling have been implicated in adhesion,15 survival, and therefore chemoresistance of leukemia16 and other tumor types.17 In chronic myelogenous leukemia, it has been suggested that activation of CXCR4 by SDF-1α within the bone marrow microenvironment could trigger integrin engagement and activation of downstream signaling molecules such as the integrin-linked kinase (ILK), further promoting leukemia cell survival.18
Integrins trigger intracellular signaling pathways by forming macromolecular complexes with different plasma membrane proteins,19 including ion channels. These are not merely bystanders, but participate in controlling integrin activation and downstream signaling.20 Determining the role of channel proteins in drug resistance assumes further importance considering the ample evidence indicating that ion channels can regulate specific stages of cancer progression and may constitute novel targets for cancer therapy.21 In particular, the voltage-dependent human ether-à-go-go-related gene 1 (hERG1) channels, whose relevance in cardiac pathophysiology is well established,22 are overexpressed in several types of human cancers.23 In these cells, hERG1 has been shown to regulate functions as different as cell proliferation, apoptosis, cell invasion, and angiogenesis by modulating several biochemical pathways. These effects seem to be often mediated by hERG1 recruitment into plasma membrane functional complexes, which generally include integrins and growth factor receptors.20,24
We determined the role of hERG1 channels in the interaction between MSCs and leukemia cells and assessed its importance in generating drug resistance in both primary clinical samples of ALL and defined cell lines.
Methods
Cell cultures
We maintained the ALL cell lines REH,25 697,26 and RS4;1127 in RPMI 1640 medium supplemented with 2mM l-glutamine, 10% bovine calf serum (HyClone). Human bone marrow–derived MSCs immortalized by telomerase reverse transcriptase transduction28 were maintained in RPMI 1640 medium supplemented with 2mM/L glutamine, 10% fetal bovine serum (HyClone), 10−6M hydrocortisone (Sigma-Aldrich), 100 U/mL penicillin, and 100 μg/mL streptomycin. MSCs were seeded in 96-well flat-bottomed plates (Costar, Corning) coated with fibronectin (1 μg/well; Sigma-Aldrich) and grown until confluence. To prepare cultures of leukemic cell lines or primary samples, we removed the media from the MSCs and washed the adherent cells 7 times with AIM-V tissue culture medium (Invitrogen). The leukemic cells were then resuspended in AIM-V medium, and 1 × 105 leukemic cells were placed on the MSC layer in each well.
Patients
Bone marrow samples from children with newly diagnosed B-cell precursor ALL (BCP-ALL) were analyzed at the Hematology-Oncology Laboratory of the Department of Pediatrics, University of Padova. Diagnosis was made according to standard cytomorphology, cytochemistry, and immunophenotypic criteria. The ethical committee of the Azienda Ospedaliera-Universitaria di Careggi approved the experiments performed.
Flow cytometry
To quantify the expression of hERG1, β1-integrin, CXCR4, CD19, CD10, and CD45 on the plasma membrane of leukemic cell lines and primary samples, we labeled the cells with the anti-hERG1 clone A12 (produced in our laboratory and sold through Enzo Life Sciences), anti-CXCR4 (2074; Abcam), anti–β1-integrin subunit clone TS2/16 (BioLegend), anti–CD10-PE (Beckman Coulter-Immunotech), anti–CD45-PE-Texas Red (Beckman Coulter-Immunotech), and anti–CD19-PE-Cy7 (Beckman Coulter-Immunotech). The cells, which had been previously washed with phosphate-buffered saline (PBS), were stained for 15 minutes at 20°C. After washing, the samples were stained for 15 minutes at 20°C with anti–mouse immunoglobulin G (IgG) fluorescein isothiocyanate (FITC) or anti–rabbit IgG FITC (Chemicon), and analyzed with an FACSCanto flow cytometer and DiVa software (Becton Dickinson). Mean fluorescence intensity ratios (MFIs) were calculated by dividing the MFI of the CXCR4, β1-integrin, or hERG1 molecule, respectively, by the MFI of the respective negative control.
Apoptosis analysis
We undertook studies with annexin V/propidium iodide (Annexin-V-FLUOS staining kit; Roche) to measure apoptosis. Leukemic cells were washed twice with PBS and then stained with anti-CD45 to exclude contamination of MSCs (supplemental Figure 3, available on the Blood Web site; see the Supplemental Materials link at the top of the online article). Cells were then resuspended in 100 μL of binding buffer and incubated with FITC-conjugated annexin V and propidium iodide. The mixture was incubated at room temperature for 15 minutes before flow cytometric analysis with an FACSCalibur flow cytometer.
Real-time quantitative polymerase chain reaction
Total RNA was extracted and the quality verified with a Bioanalyzer 2100 and the RNA 6000 Nano Assay Kit (both from Agilent Technologies). hERG1 mRNA was quantified by real-time quantitative polymerase chain reaction (RQ-PCR), with the PRISM 7700 sequence detection system (Applied Biosystems) and the SYBR Green PCR Master Mix Kit (Applied Biosystems) as reported by Pillozzi et al,29 except that the following primers, designed by Primer Express software (Applied Biosystems), were used: hERG1 forward TACTGTGACCTACACAAGATCC, reverse GATCATGTTGGTATCTCGCAG. The primers were used at a final concentration of 100nM. The GUSB gene was used as a standard reference and tested in each sample.29 The relative expression of hERG1 was calculated using a comparative threshold cycle method. The expression levels of the target genes and the reference gene were determined from the calibration curve by the following equation:
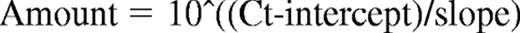
The target gene amount was then divided by the reference gene amount to obtain a normalized target value. The normalized gene target expressions were also calculated for normal CD19+ cells (calibrator). Each of the specimen-normalized gene target values was then divided by the calibrator-normalized gene target value to generate the final relative expression.
Patch-clamp recordings and data analysis
Measurements in 697 cells were performed in whole-cell mode with an Axopatch 1-D amplifier (Molecular Devices) at room temperature (20-22°C). Cells were plated onto 35-mm Petri dishes coated with polylysine (1 mg/mL) and incubated for 50 minutes at 37°C. Dishes were then placed under an inverted microscope (Nikon) and continuously perfused (at 60 μL/min) with a solution containing: NaCl 150mM, KCl 5mM, MgCl2 2mM, CaCl2 2mM, HEPES 10mM, and glucose 5mM. pH was adjusted to 7.4 with NaOH. Patch pipettes contained: K-aspartate 130mM, NaCl 10mM, MgCl2 2mM, CaCl2 4mM, EGTA-KOH 10mM, and Hepes KOH 10mM at pH 7.4. Resistance was 4-6 MΩ. Currents were filtered at 2 kHz and acquired online at 5 kHz with pClamp hardware and software (Molecular Devices). The pipette and cell capacitance and the series resistance (up to about 75%) were compensated for before each voltage-clamp protocol run. Extracellular solutions were delivered through a 9-hole (0.6-mm) remote-controlled linear positioner placed near the cell under study. Cells were maintained in our standard solution until the whole-cell configuration was obtained. Subsequently, to magnify hERG1 current (IHERG) amplitude at −120 mV, [K+]o was increased to 40mM, which brings the Nernst potential for K+ to about −30 mV. Osmolarity was kept constant by lowering [Na+]o. IHERG were isolated from the background currents by subtracting the current obtained in the presence of the specific inhibitor WAY 123,398 (2μM) from the total current. For data analysis, Clampfit 8 (Molecular Devices) and Origin 6 (Microcal Software) software were routinely used. The activation curve was calculated as illustrated in Schonherr et al.30
Immunoprecipitation and Western blotting
Protein extraction, immunoprecipitation, and Western blotting were performed essentially as in Pillozzi et al,29 and are detailed in supplemental Methods. Western blotting images were acquired with an Epson 3200 scanner, and the relative bands were analyzed by Scion Image software (Scion Corporation). The intensity of the bands was normalized to the intensity of the bands corresponding to the α-tubulin protein. The control cells ratio was set as one. The ILK assay was performed with a nonradioactive Akt kinase kit (Cell Signaling Technology) by testing the phosphorylation of the substrate (GSK-3) by Western blotting with the anti-pGSK-3 Ser9/21 antibody according to the method of Tabe et al.16 Details are reported in supplemental Methods.
Pharmacology experiments
To test drug cytotoxicity against B-ALL cells, we resuspended 1 × 105 as described in “Cell cultures.” Cell suspensions (200 μL) were placed in a 96-well flat-bottomed plate with or without bone marrow–derived MSCs. Drugs were dissolved as follows at the concentrations indicated: prednisone (Sigma-Aldrich) in ethanol:chloroform 1:1 at 50 mg/mL; doxorubicin (Amersham, GE Healthcare) vial containing 50 mg of doxorubicin chlorohydrate in NaCl, pH 3; methotrexate (Sigma-Aldrich) in 0.1M NaOH at 20 mg/mL; E4031 (Sigma-Aldrich) in distilled water at 5mM; WAY 123,398 (kindly gifted by Dr W. Spinelli, Wyeth-Ayerst Research, Princeton, NJ) in distilled water at 5mM; erythromycin (Sigma-Aldrich) in absolute ethanol at 0.1M; sertindole (kindly gifted by Lundbeck A/S, Denmark) in dimethylsulfoxide at 5mM; and R-roscovitine (Alomone Labs) in dimethylsulfoxide at 5mM. Drugs were used at the final concentrations indicated in supplemental Figure 6 and in the legends to Figures 4 and 5. Cultures were maintained for 48 hours at 37°C, 5% CO2, and 90% humidity. After incubation, cells were separated from MSCs by pipetting with ice-cold PBS and processed for apoptosis analysis as described in “Apoptosis analysis.” The LD50 value (ie, the dose that caused the apoptosis of 50% of leukemic cells) was calculated by fitting the data points with a sigmoidal curve using Origin 6 software. To determine the synergistic, additive, or antagonistic effects of the drug combinations, we used CalcuSyn software (version 2, Biosoft), which is based on the method of the combination index (CI) of Chou and Talalay. Synergy, additivity, and antagonism were defined by a CI < 1, CI = 1, or CI > 1, respectively.
RNA interference
Cells were cultured as in “Cell cultures” in 6-well cell clusters (Costar, Corning). Twenty-four hours after plating, cells were transfected with siRNAs (44858 anti-hERG1 siRNA1; 290144 anti-hERG1 siRNA2; 44762 anti-hERG1 siRNA3; Ambion; 100nM final concentration), and siRNA scramble (50nM final concentration, Silencer Negative control #1, 4611, Ambion) according to the manufacturer's instructions. After 5 hours, the medium was changed and cells were processed for pharmacology experiments.
NOD/SCID mouse assays
Experiments in NOD/SCID mice were performed at the Laboratory of Genetic Engineering for the Production of Animal Models (LIGeMA) at the Animal House of the University of Firenze. Female 5-week-old mice (Charles Rivers Laboratories) were injected via the tail vein with 10 million cells. Those inoculated with 697 cells were treated 1 week later with E4031 daily (20 mg/kg) for 2 or 4 weeks, after which peripheral blood, bone marrow, and peripheral organs (spleen and liver) were collected for analysis. Mice inoculated with REH cells were treated 1 week later with saline, E4031 (20 mg/kg), dexamethasone (15 mg/kg), or dexamethasone (15 mg/kg) + E4031 (20 mg/kg) for 5 days a week over 2 weeks, after which time bone marrow was collected for analysis.
The analysis of human cell engraftment in NOD/SCID mice and histological and immunohistochemical analyses were performed as reported in Pillozzi et al,29 and are detailed in supplemental Methods. To assess leukemic cell apoptosis in vivo, we analyzed DNA breaks in apoptotic nuclei in paraffin-embedded sections of bone marrow using the FragEL DNA fragmentation detection kit (Calbiochem), according to the manufacturer's instructions. The percentage of apoptotic cells was determined by counting terminal deoxynucleotidyltransferase-mediated dUTP nick end-labeling (TUNEL)–positive cells (epiphysis or diaphysis, 40× magnification); these quantifications were done in triplicate (3 independent counts/marrow section).
Statistical analysis
We relied on descriptive statistics (means and SE) to summarize most datasets and made statistical comparisons with the 2-tailed Student t test and ANOVA (differences with P < .05 were considered significant). Moreover, to compare hERG1 expression in patients, P values were obtained by the Mann-Whitney test.
Results
β1/hERG1/CXCR4 complex is expressed on the surface of leukemic cells and is activated by culture with MSCs
We first determined the expression of hERG1, β1-containing integrins (VLA-4 and VLA-5), and CXCR4 on leukemic cells from children with BCP-ALL and on 3 ALL cell lines (697, REH, and RS4;11). As expected, β1-integrins and CXCR4 were both expressed by 3 representative clinical samples and the cell lines (supplemental Figure 1).31,32 We found that the hERG1 transcript was overexpressed in leukemic blasts, both cell lines, and primary samples (n = 63) compared with the expression level in bone marrow CD19+ B cells (the normal counterpart of B-lineage ALL cells) and Epstein-Barr virus-infected B lymphocytes (Figure 1A). The hERG1 protein was consistently detected by flow cytometry, both in ALL cell lines (Figure 1B) and in the bone marrow of ALL patients (Figure 1C-D top panels). hERG1 expression on the plasma membrane of primary blasts was quantified by measuring MFI values (Figure 1C). hERG1 expression appeared to be correlated with that of CD10 in CD10+ BCP-ALL blasts (Figure 1D bottom panel), while virtually no signal was observed in CD10− BCP-ALL blasts (Figure 1D right bottom panel). In addition, the hERG1 transcript was lower in CD10− patients compared with CD10+ patients (supplemental Figure 2). Expression of hERG1 was detected at low levels in CD19- and CD10-positive bone marrow cells from healthy donors (Figure 1E). To determine whether the hERG1 channel expressed in ALL cells was functional, we measured whole-cell currents using the patch-clamp technique. IHERG were measured as inward currents at −120 mV after conditioning for 10 seconds at a Vm between −70 and 0 mV. Nonspecific currents were measured by applying the same stimulation protocol in the presence of the hERG1 blocker WAY 123,398 (2μM) and then subtracting the values from the total currents.30 Figure 1F illustrates the IHERG traces measured in 697 cells, along with the corresponding activation curve showing the typical biophysical features presented by hERG1 in cancer cells.33 Similar IHERG values were observed in all of the cell lines tested (697, REH, and RS4;11).
![Figure 1. Expression of hERG1 channels by primary leukemic cells and cell lines. (A) Levels of hERG1 mRNA expression in normal B lymphocytes (CD19+), Epstein-Barr virus-infected B lymphocytes, ALL cell lines (REH, RS4;11, 697), and primary samples from BCP-ALL (n = 63) patients measured by SYBR Green RQ-PCR. Levels of the hERG1 transcript were normalized to levels of the corresponding transcript in normal CD19+ cells. Mean values for hERG1 expression are reported; the box plot at the far right shows median, 25th and 75th quartile, and extreme outlier values. (B) Surface expression of hERG1 by flow cytometry in B-ALL cell lines. (C) Surface expression of hERG1 by flow cytometry in primary BCP-ALL and evaluation of the MFI value. MFI was calculated by dividing the MFI of the hERG1 molecule by the MFI of the respective negative control. (D) Surface expression of hERG1 by flow cytometry in primary leukemic cells gated on CD19+. A representative CD10+ BCP-ALL sample is shown in the left panel and a representative CD10− BCP-ALL sample is shown on the right. Top panels, plot of hERG1 expression showing both the anti-hERG1 antibody (solid line) and the control isotype labeling (dotted line). Bottom panels, dot plots relative to hERG1 expression in conjunction with that of CD10 in the leukemic cells. (E) Surface expression of hERG1 by flow cytometry in normal CD19+ CD10+ bone marrow cells. Top and bottom panels are as in panel D. (f) IHERG in 697 cells. Current traces show IHERG elicited at −120 mV, after conditioning for 10 seconds at a Vm between −70 and 0 mV (10-mV step). For clarity, only 5 current traces are shown (those obtained after conditioning at −70, −60, −40, −30, and 0 mV). IHERG was measured in [K+]o = 40mM to increase the current amplitude at −120 mV, and was isolated by subtracting the background currents from the total currents obtained in the presence of 2μM WAY 123398. The right panel shows the corresponding activation curve in a representative experiment. Black circles are peak current values measured at the indicated conditioning potential and normalized to the maximal current. Continuous line is the Boltzmann curve best fitting the experimental data. The midpoint of activation was −47 mV. These results are representative of 5 similar experiments carried out in the same cell batch, in which the average midpoint of activation was −47 ± 1.2 mV.](/view-large/figure/8010675/zh89991064740001.jpeg)
Expression of hERG1 channels by primary leukemic cells and cell lines. (A) Levels of hERG1 mRNA expression in normal B lymphocytes (CD19+), Epstein-Barr virus-infected B lymphocytes, ALL cell lines (REH, RS4;11, 697), and primary samples from BCP-ALL (n = 63) patients measured by SYBR Green RQ-PCR. Levels of the hERG1 transcript were normalized to levels of the corresponding transcript in normal CD19+ cells. Mean values for hERG1 expression are reported; the box plot at the far right shows median, 25th and 75th quartile, and extreme outlier values. (B) Surface expression of hERG1 by flow cytometry in B-ALL cell lines. (C) Surface expression of hERG1 by flow cytometry in primary BCP-ALL and evaluation of the MFI value. MFI was calculated by dividing the MFI of the hERG1 molecule by the MFI of the respective negative control. (D) Surface expression of hERG1 by flow cytometry in primary leukemic cells gated on CD19+. A representative CD10+ BCP-ALL sample is shown in the left panel and a representative CD10− BCP-ALL sample is shown on the right. Top panels, plot of hERG1 expression showing both the anti-hERG1 antibody (solid line) and the control isotype labeling (dotted line). Bottom panels, dot plots relative to hERG1 expression in conjunction with that of CD10 in the leukemic cells. (E) Surface expression of hERG1 by flow cytometry in normal CD19+ CD10+ bone marrow cells. Top and bottom panels are as in panel D. (f) IHERG in 697 cells. Current traces show IHERG elicited at −120 mV, after conditioning for 10 seconds at a Vm between −70 and 0 mV (10-mV step). For clarity, only 5 current traces are shown (those obtained after conditioning at −70, −60, −40, −30, and 0 mV). IHERG was measured in [K+]o = 40mM to increase the current amplitude at −120 mV, and was isolated by subtracting the background currents from the total currents obtained in the presence of 2μM WAY 123398. The right panel shows the corresponding activation curve in a representative experiment. Black circles are peak current values measured at the indicated conditioning potential and normalized to the maximal current. Continuous line is the Boltzmann curve best fitting the experimental data. The midpoint of activation was −47 mV. These results are representative of 5 similar experiments carried out in the same cell batch, in which the average midpoint of activation was −47 ± 1.2 mV.
Expression of hERG1 channels by primary leukemic cells and cell lines. (A) Levels of hERG1 mRNA expression in normal B lymphocytes (CD19+), Epstein-Barr virus-infected B lymphocytes, ALL cell lines (REH, RS4;11, 697), and primary samples from BCP-ALL (n = 63) patients measured by SYBR Green RQ-PCR. Levels of the hERG1 transcript were normalized to levels of the corresponding transcript in normal CD19+ cells. Mean values for hERG1 expression are reported; the box plot at the far right shows median, 25th and 75th quartile, and extreme outlier values. (B) Surface expression of hERG1 by flow cytometry in B-ALL cell lines. (C) Surface expression of hERG1 by flow cytometry in primary BCP-ALL and evaluation of the MFI value. MFI was calculated by dividing the MFI of the hERG1 molecule by the MFI of the respective negative control. (D) Surface expression of hERG1 by flow cytometry in primary leukemic cells gated on CD19+. A representative CD10+ BCP-ALL sample is shown in the left panel and a representative CD10− BCP-ALL sample is shown on the right. Top panels, plot of hERG1 expression showing both the anti-hERG1 antibody (solid line) and the control isotype labeling (dotted line). Bottom panels, dot plots relative to hERG1 expression in conjunction with that of CD10 in the leukemic cells. (E) Surface expression of hERG1 by flow cytometry in normal CD19+ CD10+ bone marrow cells. Top and bottom panels are as in panel D. (f) IHERG in 697 cells. Current traces show IHERG elicited at −120 mV, after conditioning for 10 seconds at a Vm between −70 and 0 mV (10-mV step). For clarity, only 5 current traces are shown (those obtained after conditioning at −70, −60, −40, −30, and 0 mV). IHERG was measured in [K+]o = 40mM to increase the current amplitude at −120 mV, and was isolated by subtracting the background currents from the total currents obtained in the presence of 2μM WAY 123398. The right panel shows the corresponding activation curve in a representative experiment. Black circles are peak current values measured at the indicated conditioning potential and normalized to the maximal current. Continuous line is the Boltzmann curve best fitting the experimental data. The midpoint of activation was −47 mV. These results are representative of 5 similar experiments carried out in the same cell batch, in which the average midpoint of activation was −47 ± 1.2 mV.
We next asked whether hERG1 and β1-integrin could form a macromolecular complex in ALL cells, as has been described in other tumor cell types.20,29,34 Coimmunoprecipitation experiments were performed with cells maintained under standard culture conditions (ie, in suspension in the presence of serum). An anti-β1 antibody was used to immunoprecipitate the complex, and the blotted membrane was labeled with anti-CXCR4 and anti-hERG1 antibodies. In parallel experiments, immunoprecipitation was performed with an anti-CXCR4 antibody, and the blot was labeled with anti–β1-integrin and anti-hERG1 antibodies. We found that β1-integrin, hERG1, and CXCR4 coimmunoprecipitated in all of the ALL cell lines studied (Figure 2A).

The β1/hERG1/CXCR4 complex is assembled on the plasma membrane of leukemic cells cultured on MSCs. (A) Coimmunoprecipitation (IP) of β1-integrin, hERG1, and CXCR4 from the REH, RS4;11, and 697 ALL cell lines. Left, cell lysates were immunoprecipitated with the anti–β1-integrin antibody TS2/16 and blots were probed with the anti-pan hERG1 antibody (C54), the anti-CXCR4 antibody, and the anti-β1 antibody. Right, cell lysates were immunoprecipitated with the anti-CXCR4 antibody and blots were probed with the anti-pan hERG1 antibody (C54), the anti-β1 antibody, and the anti-CXCR4 antibody. Experiments shown are representative of at least 6 independent experiments. (B) Effect of stimulation with SDF-1α, β1-integrin, and MSCs on CXCR4/β1/hERG1 complex formation. Leukemic cells were treated for 30 minutes with bovine serum albumin (BSA; 250 μg/mL), SDF-1α (50 ng/mL), the β1-integrin-activating antibody TS2/16 (20 μg/mL; left panel), or were cultured on MSCs; controls were cells treated with BSA (250 μg/mL) for 30 minutes. Experiments shown are representative of at least 4 independent experiments. (C) The 697 cell line was cultured on MSCs for 30 minutes in the absence or presence of the anti–β1-integrin-blocking antibody. Proteins were extracted and immunoprecipitated with the anti-CXCR4 antibody and the blot was sequentially labeled with anti-pan hERG1 (C54), anti–β1-integrin, or anti-CXCR4 antibody. Experiments shown are representative of at least 6 independent experiments. (D) Coimmunoprecipitation of CXCR4, hERG1, and β1-integrin from 3 primary clinical samples. Experiments were performed as described in panel A and shown are representatives of at least 4 independent experiments.
The β1/hERG1/CXCR4 complex is assembled on the plasma membrane of leukemic cells cultured on MSCs. (A) Coimmunoprecipitation (IP) of β1-integrin, hERG1, and CXCR4 from the REH, RS4;11, and 697 ALL cell lines. Left, cell lysates were immunoprecipitated with the anti–β1-integrin antibody TS2/16 and blots were probed with the anti-pan hERG1 antibody (C54), the anti-CXCR4 antibody, and the anti-β1 antibody. Right, cell lysates were immunoprecipitated with the anti-CXCR4 antibody and blots were probed with the anti-pan hERG1 antibody (C54), the anti-β1 antibody, and the anti-CXCR4 antibody. Experiments shown are representative of at least 6 independent experiments. (B) Effect of stimulation with SDF-1α, β1-integrin, and MSCs on CXCR4/β1/hERG1 complex formation. Leukemic cells were treated for 30 minutes with bovine serum albumin (BSA; 250 μg/mL), SDF-1α (50 ng/mL), the β1-integrin-activating antibody TS2/16 (20 μg/mL; left panel), or were cultured on MSCs; controls were cells treated with BSA (250 μg/mL) for 30 minutes. Experiments shown are representative of at least 4 independent experiments. (C) The 697 cell line was cultured on MSCs for 30 minutes in the absence or presence of the anti–β1-integrin-blocking antibody. Proteins were extracted and immunoprecipitated with the anti-CXCR4 antibody and the blot was sequentially labeled with anti-pan hERG1 (C54), anti–β1-integrin, or anti-CXCR4 antibody. Experiments shown are representative of at least 6 independent experiments. (D) Coimmunoprecipitation of CXCR4, hERG1, and β1-integrin from 3 primary clinical samples. Experiments were performed as described in panel A and shown are representatives of at least 4 independent experiments.
We then tested whether the complex formation was triggered by stimulation of β1-integrin or CXCR4 or perhaps both. Treatment of ALL cell lines with the CXCR4-ligand SDF-1α induced the assembly of a CXCR4/hERG1 complex (Figure 2B), but under these conditions, β1-integrin was not recruited. By contrast, when β1-integrin was stimulated by treating the cells with the TS2/16-activating antibody, in the absence or presence of SDF-1α, it was recruited to the complex in all of the cell lines studied (Figure 2B). Similar results were obtained by culturing ALL cells on MSCs (Figure 2B), suggesting that MSCs activate β1-integrin on leukemic cells while providing ligation of CXCR4 by MSC-derived SDF-1α.35 Interaction of integrins with their ligands on MSC layers was critical to the formation of the complex, which was largely disrupted by applying anti–β1-blocking antibodies (Figure 2C). Similar results were obtained by culturing primary BCP-ALL blasts on MSCs (Figure 2D). These data indicate that contact of ALL cells with MSCs results in the formation of a β1-integrin/hERG1/CXCR4 complex on the plasma membrane.
β1-integrin/hERG1/CXCR4 complex regulates MSC-induced signaling pathways: role of hERG1 channel
We next studied the signaling pathways activated by the components of the β1-integrin/hERG1/CXCR4 complex. ILK turned out to be activated after CXCR4 engagement, after culture of leukemic cells on MSCs, or both (Figure 3A). ILK activity strongly increased in leukemic cells after CXCR4 stimulation. The effect was even stronger when β1-integrin and CXCR4 were stimulated simultaneously. In all of the cell lines we have studied, the strongest activation of ILK was produced by culturing leukemic cells on MSCs (Figure 3A top panel and supplemental Figure 3a). No significant variation in the total amount of ILK was observed between cell types (Figure 3A bottom panel, and supplemental Figure 3a). ILK activation by MSCs was sustained by integrin activation, being markedly reduced in the presence of anti-β1 antibody (Figure 3A and supplemental Figure 3a). The ERK1/2 and PI3K/Akt pathways were also activated in leukemic cells cultured on MSCs (Figure 3B and supplemental Figure 3c-d). ERK1/2 phosphorylation peaked after 60 minutes, and remained high for at least 24 hours after cell seeding on MSCs (Figure 3B left panel, and E top panel). Phosphorylation of Akt increased during the first 30 minutes and then declined (Figure 3B right panel and bottom panel). Activation of both signaling pathways depended on the stimulation of β1-integrin, because it was inhibited by the blocking antibody (Figure 3B). ILK activity (Figure 3C and supplemental Figure 2b) and ERK1/2 and Akt phosphorylation (Figure 3D-E and supplemental Figure 3c-d) were strongly reduced in leukemic cells cultured on MSCs in the presence of the specific hERG1 blockers E4031 and WAY (20μM). We selected this dose because we previously demonstrated36 that low concentrations of the inhibitors (1-2μM) are not active on IHERG in protein-containing media such as those used for tissue culture. The same results were obtained with primary clinical samples (Figure 3F). For all experiments shown in Figure 3, we first determined the purity and viability of the leukemic cells recovered from culture on MSC, both in the absence and in the presence of the hERG1 inhibitor (supplemental Figure 4) and ensured that treatment with hERG1 inhibitors did not affect the expression of β1 and the secretion of SDF-1α from MSCs (supplemental Figure 5).

The β1-integrin/hERG1/CXCR4 complex regulates MSC-induced signaling. (A) Western blot of 697 cells treated for 30 minutes with BSA (250 μg/mL), SDF-1α (50 ng/mL), the β1-integrin-activating antibody TS2/16 (20 μg/mL), or cultured on MSCs with or without the anti–β1-integrin-blocking antibody. Total cell lysates were immunoprecipitated with a polyclonal ILK antibody, and a kinase assay was performed with the synthetic GSK3 protein as a substrate. Phosphorylated substrates were detected by Western blot with the anti–pGSK-3 Ser9/21 antibody. Densitometric measurements relative to the blot reported are shown on the bottom. Experiments reported in the panel are representative of at least 4 independent experiments. (B) Western blot of pERK1/2 (left panel) and pAkt (right panel) in 697 cells after coculture for 30 or 60 minutes with MSCs in the absence or presence of anti–β1-integrin-blocking antibody. The membrane was then reprobed with an anti-ERK1/2 or anti-Akt antibody and anti-tubulin antibody. Densitometric measurements relative to the blot reported in the panel are shown on the bottom. Experiments shown are representative of at least 4 independent experiments. (C) 697 cells were cultured on MSCs for 30 minutes with or without an hERG1 blocker (WAY or E4031, both at 20μM). Cell lysates were immunoprecipitated and processed as in panel A. Densitometric measurements relative to the blot reported are shown on the bottom. Experiments shown are representative of at least 4 independent experiments. (D) Western blot of the levels of pERK1/2 and pAkt in 697 cells after coculture with MSCs for 30 minutes, 60 minutes, or 24 hours with or without the hERG1 inhibitor E4031 (20μM). Total levels of ERK1/2 and Akt and tubulin proteins are shown. Densitometric measurements relative to the blot reported are shown on the bottom (white, −E4031; gray, +E4031). Experiments shown are averaged in panel E and are representative of 5 independent experiments. (E) pERK1/2 (top panel) and pAkt (bottom panel) in leukemic cells cultured on MSCs in the absence (dashed line) or in the presence of hERG1 inhibitors (solid black line) over 24 hours; densitometric measurements shown represent the mean of 5 different experiments. *P < .05. (F) Western blot of pERK and pAkt levels in primary clinical samples of BCP-ALL after coculture with MSCs for 30 or 60 minutes in the absence or presence of the hERG1 inhibitor E4031 (20μM). Total levels of ERK1/2 and Akt proteins and tubulin are shown in the bottom panels.
The β1-integrin/hERG1/CXCR4 complex regulates MSC-induced signaling. (A) Western blot of 697 cells treated for 30 minutes with BSA (250 μg/mL), SDF-1α (50 ng/mL), the β1-integrin-activating antibody TS2/16 (20 μg/mL), or cultured on MSCs with or without the anti–β1-integrin-blocking antibody. Total cell lysates were immunoprecipitated with a polyclonal ILK antibody, and a kinase assay was performed with the synthetic GSK3 protein as a substrate. Phosphorylated substrates were detected by Western blot with the anti–pGSK-3 Ser9/21 antibody. Densitometric measurements relative to the blot reported are shown on the bottom. Experiments reported in the panel are representative of at least 4 independent experiments. (B) Western blot of pERK1/2 (left panel) and pAkt (right panel) in 697 cells after coculture for 30 or 60 minutes with MSCs in the absence or presence of anti–β1-integrin-blocking antibody. The membrane was then reprobed with an anti-ERK1/2 or anti-Akt antibody and anti-tubulin antibody. Densitometric measurements relative to the blot reported in the panel are shown on the bottom. Experiments shown are representative of at least 4 independent experiments. (C) 697 cells were cultured on MSCs for 30 minutes with or without an hERG1 blocker (WAY or E4031, both at 20μM). Cell lysates were immunoprecipitated and processed as in panel A. Densitometric measurements relative to the blot reported are shown on the bottom. Experiments shown are representative of at least 4 independent experiments. (D) Western blot of the levels of pERK1/2 and pAkt in 697 cells after coculture with MSCs for 30 minutes, 60 minutes, or 24 hours with or without the hERG1 inhibitor E4031 (20μM). Total levels of ERK1/2 and Akt and tubulin proteins are shown. Densitometric measurements relative to the blot reported are shown on the bottom (white, −E4031; gray, +E4031). Experiments shown are averaged in panel E and are representative of 5 independent experiments. (E) pERK1/2 (top panel) and pAkt (bottom panel) in leukemic cells cultured on MSCs in the absence (dashed line) or in the presence of hERG1 inhibitors (solid black line) over 24 hours; densitometric measurements shown represent the mean of 5 different experiments. *P < .05. (F) Western blot of pERK and pAkt levels in primary clinical samples of BCP-ALL after coculture with MSCs for 30 or 60 minutes in the absence or presence of the hERG1 inhibitor E4031 (20μM). Total levels of ERK1/2 and Akt proteins and tubulin are shown in the bottom panels.
Thus, the β1-integrin/hERG1/CXCR4 complex participates in the interaction between MSCs and ALL cells, and the activity of hERG1 channels inside the complex is necessary for the complex to regulate the integrin-mediated signaling.
hERG1 channel function drives MSC-induced chemoresistance of leukemic cells
When cultured on MSCs, leukemic cells are protected from the apoptosis induced by chemotherapy.5-11 We tested this effect and the possible role of hERG1. In the first set of experiments, leukemic cells were exposed to doxorubicin, prednisone, or methotrexate, drugs commonly used to treat ALL, in the presence or absence of the hERG1-inhibitor E4031 both in suspension and on MSCs. We estimated the concentration of each drug that caused 50% cytotoxicity with or without the LD50 concentration of E4031 (20μM) and analyzed synergistic interactions. LD50 values are reported in Table 1, while the complete dose-response curves, along with the CI values of the combinations between chemotherapeutic drugs and E4031, are shown in supplemental Figure 6. As expected, the LD50 value of each chemotherapeutic drug increased in cultures containing MSCs. The addition of LD50 concentrations of E4031 lowered the LD50 of all drugs regardless of whether MSCs were present. Moreover, E4031 was synergic with the chemotherapeutic drugs at concentrations equal to or higher than the LD50 dose. The differences in both parameters (LD50 and CI) were especially evident in MSC-supported cultures, in which the addition of E4031 essentially abrogated the protective effect of MSCs. Interestingly, E4031 did not affect the viability of peripheral blood lymphocytes derived from healthy donors (supplemental Table 1). These results suggest that E4031 potentiates the proapoptotic effect of chemotherapeutic drugs, and that this effect is increased when leukemic cells are in contact with MSCs. Even when used at concentrations below LD50, E4031 enhanced the proapoptotic effects of doxorubicin, prednisone, and methotrexate, an effect that was more prominent in the presence of MSCs (Figure 4 and supplemental Figure 7). Moreover, the synergic interaction between chemotherapeutic drugs and E4031 was more evident with low doses (5μM) of E4031, and on MSCs (Figure 4 and Table 2). This occurred in both B-ALL cell lines and primary BCP-ALL samples (Figure 4C-D, supplemental Figure 7c, and Table 2).
LD50 values for doxorubicin, prednisone, and methotrexate in 697 cells cultured with or without MSCs (suspension) in combination or not with the LD50 dose of the hERG1 inhibitor E4031 in B-ALL*
| Suspension | ΜSC | |
|---|---|---|
| Doxorubicin | 0.13 ± 0.04 μg/mL | 0.42 ± 0.06 μg/mL |
| Doxorubicin + E4031 | 0.08 ± 0.04 μg/mL | 0.05 ± 0.01 μg/mL |
| Prednisone | 4.96 ± 1.01 μΜ | 23.82 ± 8.63 μΜ |
| Prednisone + E4031 | 3.77 ± 0.93 μΜ | 3.48 ± 0.97 μΜ |
| Μethotrexate | 2.16 ± 1.33 μΜ | 15.04 ± 2.56 μΜ |
| Μethotrexate + E4031 | 1.20 ± 0.09 μΜ | 2.12 ± 0.65 μΜ |
| E4031 | 22.01 ± 2.34 μΜ | 75.32 ± 5.68 μΜ |
| Roscovitine | 29.13 ± 3.67 μΜ | 34.57 ± 3.51 μΜ |
| Suspension | ΜSC | |
|---|---|---|
| Doxorubicin | 0.13 ± 0.04 μg/mL | 0.42 ± 0.06 μg/mL |
| Doxorubicin + E4031 | 0.08 ± 0.04 μg/mL | 0.05 ± 0.01 μg/mL |
| Prednisone | 4.96 ± 1.01 μΜ | 23.82 ± 8.63 μΜ |
| Prednisone + E4031 | 3.77 ± 0.93 μΜ | 3.48 ± 0.97 μΜ |
| Μethotrexate | 2.16 ± 1.33 μΜ | 15.04 ± 2.56 μΜ |
| Μethotrexate + E4031 | 1.20 ± 0.09 μΜ | 2.12 ± 0.65 μΜ |
| E4031 | 22.01 ± 2.34 μΜ | 75.32 ± 5.68 μΜ |
| Roscovitine | 29.13 ± 3.67 μΜ | 34.57 ± 3.51 μΜ |
The 697 cell line was cultured with or without MSCs (suspension) and exposed to increasing concentrations of doxorubicin, prednisone or methotrexate for 48 hours. The percentage of annexin V+/PI− cells was measured. LD50 values were evaluated by nonlinear regression analysis using Origin 6 software (Microcal Software). Original dose-response curves are reported in supplemental Figure 6.

Effect of E4031 on chemotherapy-induced apoptosis in different culture conditions. The 697 cell line and a representative BCP-ALL primary sample (see also supplemental Figure 7) were cultured with or without MSCs (suspension) and exposed to LD50 doses of either doxorubicin (0.1 μg/mL), prednisone (5μM), or methotrexate (1.5μM), with or without the LD50 dose (20μM) or a subcytotoxic dose (5μM) of the hERG1 inhibitor E4031 for 48 hours. The percentage of annexin V+/propidium iodide− cells was measured. Values are means ± SD of 6 independent experiments (for the 697 cell line, 2 experiments for the BCP-ALL primary sample), each performed in quadruplicate (for the 697 cell line, in triplicate for the BCP-ALL primary sample). (A) 697 B-ALL cell line in the presence of 20μM E4031. (B) 697 B-ALL cell line in the presence of 5μM E4031. (C) BCP-ALL primary sample (case 4, see Figure 5 and supplemental Table 3) in the presence of 20μM E4031. (D) BCP-ALL primary sample (case 4, see Figure 5 and supplemental Table 3) in the presence of 5μM E4031. The values of the untreated controls are reported as dotted bars in each graph. Original values are: 697 (suspension): 3.5% ± 0.9% (n = 6); 697 (MSC): 2.8% ± 1% (n = 6); BCP-ALL(4; suspension): 11.1% ± 3.0% (n = 2); and BCP-ALL(4; MSC) 8.2% ± 2.9% (n = 2).
Effect of E4031 on chemotherapy-induced apoptosis in different culture conditions. The 697 cell line and a representative BCP-ALL primary sample (see also supplemental Figure 7) were cultured with or without MSCs (suspension) and exposed to LD50 doses of either doxorubicin (0.1 μg/mL), prednisone (5μM), or methotrexate (1.5μM), with or without the LD50 dose (20μM) or a subcytotoxic dose (5μM) of the hERG1 inhibitor E4031 for 48 hours. The percentage of annexin V+/propidium iodide− cells was measured. Values are means ± SD of 6 independent experiments (for the 697 cell line, 2 experiments for the BCP-ALL primary sample), each performed in quadruplicate (for the 697 cell line, in triplicate for the BCP-ALL primary sample). (A) 697 B-ALL cell line in the presence of 20μM E4031. (B) 697 B-ALL cell line in the presence of 5μM E4031. (C) BCP-ALL primary sample (case 4, see Figure 5 and supplemental Table 3) in the presence of 20μM E4031. (D) BCP-ALL primary sample (case 4, see Figure 5 and supplemental Table 3) in the presence of 5μM E4031. The values of the untreated controls are reported as dotted bars in each graph. Original values are: 697 (suspension): 3.5% ± 0.9% (n = 6); 697 (MSC): 2.8% ± 1% (n = 6); BCP-ALL(4; suspension): 11.1% ± 3.0% (n = 2); and BCP-ALL(4; MSC) 8.2% ± 2.9% (n = 2).
CI values for doxorubicin, prednisone, and methotrexate (at the LD50 dose) in combination with the hERG1 inhibitor E4031 (at both 20μM and 5μM) in 697 and primary BCP-ALL cells, BCP-ALL(4) and BCP-ALL(3), cultured with or without MSCs (suspension)*
| Suspension | P (suspension vs MSC) | ΜSC | P (E4031 20μM vs E4031 5μM) | |
|---|---|---|---|---|
| 697 cell line | ||||
| Doxorubicin + E4031 20μM | > 1 | 0.002 | 0.368 ± 0.037 | 0.039 |
| Doxorubicin + E4031 5μM | 0.605 ± 0.082 | 0.037 | 0.294 ± 0.020 | |
| Prednisone + E4031 20μM | > 1 | 0.041 | 0.722 ± 0.112 | 0.002 |
| Prednisone + E4031 5μM | 0.887 ± 0.103 | 0.0001 | 0.298 ± 0.075 | |
| Methotrexate + E4031 20μM | > 1 | 0.003 | 0.317 ± 0.081 | 0.021 |
| Methotrexate + E4031 5μM | 0.563 ± 0.096 | 0.023 | 0.224 ± 0.076 | |
| BCP-ALL (4) | ||||
| Doxorubicin + E4031 20μM | > 1 | 0.008 | 0.679 ± 0.087 | 0.003 |
| Doxorubicin + E4031 5μM | > 1 | 0.001 | 0.345 ± 0.033 | |
| Prednisone + E4031 20μM | > 1 | > 1 | 0.042 | |
| Prednisone + E4031 5μM | > 1 | 0.043 | 0.854 ± 0.096 | |
| Methotrexate + E4031 20μM | > 1 | 0.002 | 0.446 ± 0.081 | 0.022 |
| Methotrexate + E4031 5μM | > 1 | 0.000 | 0.203 ± 0.073 | |
| BCP-ALL (3) | ||||
| Doxorubicin + E4031 20μM | 0.865 ± 0.105 | 0.036 | 0.487 ± 0.086 | 0.031 |
| Doxorubicin + E4031 5μM | 0.824 ± 0.043 | 0.025 | 0.398 ± 0.075 | |
| Prednisone + E4031 20μM | > 1 | > 1 | 0.004 | |
| Prednisone + E4031 5μM | > 1 | 0.037 | 0.876 ± 0.056 | |
| Methotrexate + E4031 20μM | > 1 | 0.005 | 0.865 ± 0.056 | |
| Methotrexate + E4031 5μM | > 1 | 0.033 | 0.849 ± 0.021 |
| Suspension | P (suspension vs MSC) | ΜSC | P (E4031 20μM vs E4031 5μM) | |
|---|---|---|---|---|
| 697 cell line | ||||
| Doxorubicin + E4031 20μM | > 1 | 0.002 | 0.368 ± 0.037 | 0.039 |
| Doxorubicin + E4031 5μM | 0.605 ± 0.082 | 0.037 | 0.294 ± 0.020 | |
| Prednisone + E4031 20μM | > 1 | 0.041 | 0.722 ± 0.112 | 0.002 |
| Prednisone + E4031 5μM | 0.887 ± 0.103 | 0.0001 | 0.298 ± 0.075 | |
| Methotrexate + E4031 20μM | > 1 | 0.003 | 0.317 ± 0.081 | 0.021 |
| Methotrexate + E4031 5μM | 0.563 ± 0.096 | 0.023 | 0.224 ± 0.076 | |
| BCP-ALL (4) | ||||
| Doxorubicin + E4031 20μM | > 1 | 0.008 | 0.679 ± 0.087 | 0.003 |
| Doxorubicin + E4031 5μM | > 1 | 0.001 | 0.345 ± 0.033 | |
| Prednisone + E4031 20μM | > 1 | > 1 | 0.042 | |
| Prednisone + E4031 5μM | > 1 | 0.043 | 0.854 ± 0.096 | |
| Methotrexate + E4031 20μM | > 1 | 0.002 | 0.446 ± 0.081 | 0.022 |
| Methotrexate + E4031 5μM | > 1 | 0.000 | 0.203 ± 0.073 | |
| BCP-ALL (3) | ||||
| Doxorubicin + E4031 20μM | 0.865 ± 0.105 | 0.036 | 0.487 ± 0.086 | 0.031 |
| Doxorubicin + E4031 5μM | 0.824 ± 0.043 | 0.025 | 0.398 ± 0.075 | |
| Prednisone + E4031 20μM | > 1 | > 1 | 0.004 | |
| Prednisone + E4031 5μM | > 1 | 0.037 | 0.876 ± 0.056 | |
| Methotrexate + E4031 20μM | > 1 | 0.005 | 0.865 ± 0.056 | |
| Methotrexate + E4031 5μM | > 1 | 0.033 | 0.849 ± 0.021 |
Original data are from Figure 4 and supplemental Figure 7c. CI values were calculated using CalcuSyn software Version 2 (Biosoft). span lang=IT
CI > 1, antagonisms; CI = 1, additivity; CI < 1, synergy. The statistical analysis was performed using the Student t test comparing either CI values in suspension versus CI values on MSC, for each drug combination, or CI values in the presence of 20mM E4031 versus CI values in the presence of 5μM E4031. Only statistically significant P values are reported.
To determine whether these results were a unique effect of E4031 or if they extended to other hERG1 inhibitors, we tested the effects of another specific hERG1 blocker (WAY), as well as other compounds known to block hERG1 with different biophysical mechanisms: sertindole (an antipsychotic), erythromycin (an antibiotic), and R-roscovitine (a cyclin-dependent kinase inhibitor).37-43 When added to cultures of leukemic cells on MSCs together with doxorubicin, each of these blockers suppressed MSC-induced chemoprotection on leukemic cells (Figure 5A). Because hERG1 blockers could also affect other targets, we decreased hERG1 expression on the plasma membrane of leukemic cells by RNA interference (Figure 5B). This abrogated the susceptibility to MSC-induced chemoprotection against doxorubicin and prevented any further effect of E4031. These results indicate that hERG1 plays a role in the MSC chemoprotective effect, and that its suppression by hERG1 inhibitors is caused by hERG1 channel blockade.

Effects of hERG1 blockers on MSC-induced chemoresistance of leukemic cells. (A) The 697 cell line was cultured with or without MSCs and exposed to doxorubicin (0.1 μg/mL) with or without the hERG1 inhibitors WAY (20μM), sertindole (1μM), erythromycin (100μM), and R-roscovitine (20μM). The percentage of annexin V+/propidium iodide− cells was evaluated after 48 hours of culture. Values are means ± SD of 3 experiments, each performed in quadruplicate. (B) 697 cells were treated with siRNA hERG1 and siRNA scramble and then cultured with or without MSCs and exposed to doxorubicin (0.1 μg/mL) with or without E4031 (20μM). The percentage of annexin V+/propidium iodide− cells was evaluated after 48 hours of culture. Values in all panels are means ± SD of 3 experiments, each carried out in triplicate. Inset, flow cytometric analysis of hERG1 surface expression in 697 cells transfected with siRNA hERG1 (bottom) and transfected with siRNA scramble control (top). (C) Primary leukemic cells (n = 6) were exposed to doxorubicin (0.1 μg/mL) with or without the hERG1 inhibitors E4031 (20μM), WAY (20μM), and erythromycin (100μM). The percentage of annexin V+/propidium iodide− cells was evaluated after 48 hours of culture. MFI values of hERG1 are reported in the top right corner of each panel. Values in all panels are means ± SD of triplicate experiments.
Effects of hERG1 blockers on MSC-induced chemoresistance of leukemic cells. (A) The 697 cell line was cultured with or without MSCs and exposed to doxorubicin (0.1 μg/mL) with or without the hERG1 inhibitors WAY (20μM), sertindole (1μM), erythromycin (100μM), and R-roscovitine (20μM). The percentage of annexin V+/propidium iodide− cells was evaluated after 48 hours of culture. Values are means ± SD of 3 experiments, each performed in quadruplicate. (B) 697 cells were treated with siRNA hERG1 and siRNA scramble and then cultured with or without MSCs and exposed to doxorubicin (0.1 μg/mL) with or without E4031 (20μM). The percentage of annexin V+/propidium iodide− cells was evaluated after 48 hours of culture. Values in all panels are means ± SD of 3 experiments, each carried out in triplicate. Inset, flow cytometric analysis of hERG1 surface expression in 697 cells transfected with siRNA hERG1 (bottom) and transfected with siRNA scramble control (top). (C) Primary leukemic cells (n = 6) were exposed to doxorubicin (0.1 μg/mL) with or without the hERG1 inhibitors E4031 (20μM), WAY (20μM), and erythromycin (100μM). The percentage of annexin V+/propidium iodide− cells was evaluated after 48 hours of culture. MFI values of hERG1 are reported in the top right corner of each panel. Values in all panels are means ± SD of triplicate experiments.
The effect of blocking hERG1 with E4031, WAY, and erythromycin was also tested on leukemic cells from 6 cases of primary BCP-ALL cultured in the absence or presence of MSCs (Figure 5C). All of these drugs prevented the MSC-induced chemoprotection against doxorubicin, although to an extent that varied somewhat between drugs and between specific leukemia cases. This effect was correlated with the amount of hERG1 protein expressed on the plasma membrane of leukemic cells (Figure 5C and supplemental Figure 8). We conclude that hERG1 activity regulates MSC-induced drug resistance, which in turn can be significantly overcome by administering an hERG1 inhibitor.
Effects of hERG1 blockers in vivo
We next determined whether hERG1 inhibitors could improve the outcome of treatment in murine models of ALL. In the first set of experiments (Figure 6A-C), 697 cells were inoculated in immunodeficient (NOD-SCID) mice. The mice were subsequently treated daily with E4031 (20 mg/kg) for 2 weeks starting 1 week after the inoculum. At the end of treatment, some of the mice were killed, and the degree of bone marrow, peripheral blood, and extramedullary organ invasion by ALL cells was quantified (see “Methods” section for details). The remaining mice were used to determine overall survival. E4031 treatment significantly reduced the leukemia burden and the infiltration of liver and spleen by leukemic cells (Figure 6B-C), with a significant prolongation of overall survival (Figure 6C).

hERG1 blockers can overcome MSC-induced chemoresistance in vivo. NOD-SCID mice (n = 24) were inoculated with 697 cells on day 0 and treated with E4031 daily (20 mg/kg) for 2 weeks beginning 1 week after inoculation. At the end of the treatment, mice were killed, and peripheral blood (PB), bone marrow (BM), and peripheral organs (spleen, liver) were collected. (A) Leukemia engraftment in peripheral blood and bone marrow was quantified by determining the percentage of human CD45-positive (hCD45+) cells versus mouse CD45-positive (mCD45+) cells by flow cytometry. (B-C) Immunohistochemical staining with anti-hMHCI antibody of the liver and spleen of mice inoculated with the 697 cell line, and either treated (right) or not (left) with E4031. Arrows indicate foci of leukemic blasts. Histograms quantifying the extent of leukemic cell invasion (by anti-hMHCI staining) are shown on the right. The presence and extent of leukemia blast invasion is given as the area positive for anti-hMHCI staining calculated with Leica DC Viewer software at 40× magnification. Images were acquired on a DM 4000B microscope with a DFC 320 camera (Leica Microsystems; PL Fluotar 40×/0.70 objective) Scale bar = 100 μm. (D) Kaplan-Meier plots of overall survival among mice treated with saline only (n = 12) or E4031 (n = 12) after inoculation of 697 leukemic cells.
hERG1 blockers can overcome MSC-induced chemoresistance in vivo. NOD-SCID mice (n = 24) were inoculated with 697 cells on day 0 and treated with E4031 daily (20 mg/kg) for 2 weeks beginning 1 week after inoculation. At the end of the treatment, mice were killed, and peripheral blood (PB), bone marrow (BM), and peripheral organs (spleen, liver) were collected. (A) Leukemia engraftment in peripheral blood and bone marrow was quantified by determining the percentage of human CD45-positive (hCD45+) cells versus mouse CD45-positive (mCD45+) cells by flow cytometry. (B-C) Immunohistochemical staining with anti-hMHCI antibody of the liver and spleen of mice inoculated with the 697 cell line, and either treated (right) or not (left) with E4031. Arrows indicate foci of leukemic blasts. Histograms quantifying the extent of leukemic cell invasion (by anti-hMHCI staining) are shown on the right. The presence and extent of leukemia blast invasion is given as the area positive for anti-hMHCI staining calculated with Leica DC Viewer software at 40× magnification. Images were acquired on a DM 4000B microscope with a DFC 320 camera (Leica Microsystems; PL Fluotar 40×/0.70 objective) Scale bar = 100 μm. (D) Kaplan-Meier plots of overall survival among mice treated with saline only (n = 12) or E4031 (n = 12) after inoculation of 697 leukemic cells.
In a second set of experiments, we tested the effects of combined treatment with E4031 and dexamethasone on REH cells, which have been reported to be relatively resistant to corticosteroids (in vitro LD50 =‖;> 10μM).44 Mice were treated for 2 weeks with dexamethasone, E4031, or both (Figure 7). Treatment with dexamethasone and E4031 in combination nearly abolished bone marrow engraftment, while producing marked apoptosis (Figure 7A) and strongly reducing the proportion of leukemic cells in the peripheral blood (Figure 7B) and leukemia infiltration of extramedullary sites (Figure 7C-D). These effects were significantly superior to those obtained by treatment with either dexamethasone alone or E4031.

Effects of combined treatment with E4031 and dexamethasone in vivo. (A) NOD-SCID mice (n = 16, 4 for each group) were inoculated with REH cells on day 0, and 1 week after inoculation were treated with: (i) saline, (ii) E4031 (20 mg/kg), (iii) dexamethasone (15 mg/kg), or intravenous dexamethasone (15 mg/kg) plus E4031 (20 mg/kg) for 2 weeks. At the end of treatment, mice were killed and bone marrow, spleens, and livers were collected. Left panel, bone marrow engraftment in control, dexamethasone-, dexamethasone + E4031-, and E4031-treated mice, quantified by determining the percentage of hCD45+ versus mCD45+ cells by flow cytometry. Right panel, FragEL staining of bone marrow from mice inoculated with REH cells and treated as in panel A. Results show the number of TUNEL-positive leukemic cells per microscopic field (epiphysis or diaphysis, 40× magnification). The data are means ± SD of triplicate experiments. (B) Peripheral blood invasion in control, dexamethasone-, dexamethasone + E4031-, and E4031-treated mice, quantified by determining the percentage of hCD45+ versus mCD45+ cells by flow cytometry. (C) Extent of leukemic cell invasion in the spleen (left) and in the liver (right) of mice inoculated with REH cells and treated as in panel A. The extent of leukemic cell infiltration was calculated by anti-hMHCI staining measured with Leica DC Viewer software at 40×. Images were acquired on a Leica DM 4000B microscope with a Leica DFC 320 camera (Leica Microsystems; PL Fluotar 40×/0.70 objective) *P < .05. (D) Immunohistochemical staining with an anti-hMHCI antibody of the spleens of mice inoculated with the REH cell line and treated as in panel A. Scale bar = 50 μm. Inset, magnification of images reported on the left. *P < .05; **P < .01; ***P < .001
Effects of combined treatment with E4031 and dexamethasone in vivo. (A) NOD-SCID mice (n = 16, 4 for each group) were inoculated with REH cells on day 0, and 1 week after inoculation were treated with: (i) saline, (ii) E4031 (20 mg/kg), (iii) dexamethasone (15 mg/kg), or intravenous dexamethasone (15 mg/kg) plus E4031 (20 mg/kg) for 2 weeks. At the end of treatment, mice were killed and bone marrow, spleens, and livers were collected. Left panel, bone marrow engraftment in control, dexamethasone-, dexamethasone + E4031-, and E4031-treated mice, quantified by determining the percentage of hCD45+ versus mCD45+ cells by flow cytometry. Right panel, FragEL staining of bone marrow from mice inoculated with REH cells and treated as in panel A. Results show the number of TUNEL-positive leukemic cells per microscopic field (epiphysis or diaphysis, 40× magnification). The data are means ± SD of triplicate experiments. (B) Peripheral blood invasion in control, dexamethasone-, dexamethasone + E4031-, and E4031-treated mice, quantified by determining the percentage of hCD45+ versus mCD45+ cells by flow cytometry. (C) Extent of leukemic cell invasion in the spleen (left) and in the liver (right) of mice inoculated with REH cells and treated as in panel A. The extent of leukemic cell infiltration was calculated by anti-hMHCI staining measured with Leica DC Viewer software at 40×. Images were acquired on a Leica DM 4000B microscope with a Leica DFC 320 camera (Leica Microsystems; PL Fluotar 40×/0.70 objective) *P < .05. (D) Immunohistochemical staining with an anti-hMHCI antibody of the spleens of mice inoculated with the REH cell line and treated as in panel A. Scale bar = 50 μm. Inset, magnification of images reported on the left. *P < .05; **P < .01; ***P < .001
Discussion
Our results point to a novel mechanism by which mesenchymal cells in the bone marrow microenvironment protect leukemic cells from chemotherapy. Central to this protection is a plasma membrane complex on leukemic cells consisting of hERG1 K+ channels, the β1-integrin subunit, and the SDF-1α receptor CXCR4. The role of hERG1 seems critical, because hERG1 inhibitors abrogate the protective effect of MSCs and enhance the cytotoxicity of drugs commonly used to treat leukemia. The finding that different pharmacological categories of hERG1 inhibitors exert the same effect reinforces the notion that hERG1 function is crucial for chemoprotection to be sustained. Finally, because some of the hERG1 blockers that proved effective in this study are readily available, our results suggest clinical testing of treatment strategies that include hERG1 inhibition.
The chemokine SDF-1α secreted by MSCs and its receptor CXCR4 are known to be critical mediators of the interaction between MSCs and leukemic cells.14 Adhesion between these cell types is consolidated by engagement of the integrin receptors expressed onto leukemic cells, typically α5β1 (VLA-5) and α4β1 (VLA-4), which provide an additional signaling mechanism by interacting with extracellular matrix proteins on the MSCs.10 Studies in acute myeloid leukemia,5 chronic lymphocytic leukemia,6-8 and chronic myeloid leukemia9 have demonstrated that antiapoptotic proteins activated by the MSC-triggered intracellular pathways confer a survival advantage to leukemic cells.10 In myeloid leukemias,16 these signals are most likely centered on the integrin-dependent activation of ILK, with ensuing recruitment of the MAPK and the PI3K/Akt pathways. Consistently, our data on ALL indicate that MSCs trigger the formation of a plasma membrane macromolecular complex that includes β1-containing integrins, the SDF-1α receptor CXCR4, and hERG1 channels. This complex is not found on mature normal B lymphocytes, perhaps explaining the loss of SDF-1α responsiveness during B-cell maturation,45,46 nor on MSCs (supplemental Figure 4a). In leukemic blasts, such a complex activates signaling pathways downstream to β1-integrin activation. These pathways include ILK, which stimulates phosphorylation of ERK1/2 MAP kinases and Akt. Activation of these signals, particularly that of Akt, produces antiapoptotic effects through phosphorylation of the substrates that directly or indirectly regulate the apoptotic machinery.47 This raises the possibility that the β1-integrin/hERG1/CXCR4 complex that we identified on leukemic lymphoblasts plays a similar regulatory role in the development of chemoresistance in myeloid leukemias (Pillozzi, unpublished results). On the plasma membrane of leukemic cells, hERG1 channels are found both as single proteins (as is also the case in heart myocytes) or in association with integrins within multiprotein complexes. Strictly speaking, our results do not distinguish between the effects produced by the channels associated with the complex and those that are not. Distinguishing the relative roles of these hERG1 populations will require further experiments with methods capable of distinguishing associated and nonassociated channels. Nonetheless, basing on previous results of ours,24 we favor the hypothesis that most of the effects described in the present paper depend on the channels recruited by the membrane complex. In fact, the fraction of hERG1 channels that have a significant functional activity (ie, reside in the open state) in tumor cells is mainly composed of the channels complexed with integrins.24 Integrin activation depends on hERG1 channel activity, because it is severely impaired by treatment with hERG1-specific blockers. Because these channels are not expressed by MSC cells, it is very unlikely that hERG1 blockade affects SDF-1α secretion or β1-integrin expression on these stromal cells, and this was formally tested in the experiments shown in supplemental Figure 4b and c. Finally, recent evidence indicates that hERG1 channels can regulate the SDF-1α-mediated migration in leukemic cells.48 Our results provide new insights into this process, indicating that the functions of CXCR4, integrins, and hERG1 tightly cooperate within a multiprotein membrane complex.
Leukemic cells are known to be protected from chemotherapy by MSCs,5-11 and hERG1 channel function appears to be important for this effect. Indeed, blocking hERG1 significantly shifted the LD50 values of 3 drugs commonly used in induction therapy of pediatric B-ALL (doxorubicin, prednisone, and methotrexate), particularly when cells were cultured on MSCs, and ALL cells with down-regulated hERG1 by RNA interference lost their susceptibility to MSC-mediated protection. Thus, agents that exclusively target hERG1 may be adequate to overcome MSC-induced drug resistance. In agreement with this idea, treating our murine models with hERG1 blockers significantly increased the rate of leukemic cell apoptosis in bone marrow and reduced leukemic infiltration of peripheral organs. Finally, in addition to class III antiarrhythmics such as E4031 and WAY,49 we found that other compounds with potent anti-hERG1 effects also exert antileukemic activity. These results present interesting implications for treatment. In fact, sertindole and erythromycin,40,41 unlike other hERG blockers, present scarce,50 antiarrhythmic effects and therefore would be promising candidates for inclusion in clinical trials.38,50
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
We thank Dr A. Lippi for believing in this research and for supporting us with valuable suggestions; Prof A. Biondi and Dr G. Gaipa for useful suggestions; H. Lundbeck A/S (Copenhagen, Denmark) for providing sertindole; and Prof E. Mini and Dr I. Landini (Department of Pharmacology, University of Firenze) for introducing us to CalcuSyn software.
This work was supported by grants from the Associazione Genitori contro le Leucemie e Tumori Infantili Noi per Voi, Associazione Italiana per la Ricerca sul Cancro, Istituto Toscano Tumori and PRIN 2008 to A.A.; from the Fondazione Città della Speranza and PRIN 2007 to B.G.; from the Ente Cassa di Risparmio di Firenze to the Dipartimento di Patologia e Oncologia Sperimentali, Università di Firenze; and from the University of Milano Bicocca (Fondi di Ateneo per la Ricerca) to A.B.
Authorship
Contribution: S.P. designed and performed research; M.M., B.A., O.C., E.D., E.C., A. Amedei, M.V., and M.D. performed research; D.C. interpreted data and contributed to writing the paper; A. Arcangeli designed research and wrote the paper; G.B. contributed primary samples; and A.B. analyzed and interpreted data and contributed to writing the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Annarosa Arcangeli, MD, PhD, Department of Experimental Pathology and Oncology, University of Florence, Viale G.B. Morgagni, 50, 50134 Firenze, Italy; e-mail: annarosa.arcangeli@unifi.it.
