

Abstract
Abstract 264
Splenic marginal zone lymphoma (SMZL) is one of few B-cell lymphoma types that remain orphan of molecular lesions in cancer related genes. Detection of active NF-κB signaling by immunohistochemical assays for nuclear NFKB1 (p50) and NFKB2 (p52) in 14/24 (58%) SMZL prompted the investigation of NF-κB molecular alterations in SMZL (Fig.1A). To this aim, NF-κB pathway genes (n=20) were screened in 101 SMZL for mutations by Sanger sequencing and for copy number abnormalities (CNAs) by FISH and SNP array (GeneChip Human Mapping 250K NspI, Affymetrix). Mutations targeting multiple NF-κB genes were detected in 20/101 (20%) SMZL. Mutations affected key regulators of both canonical (IKBKB, TNFAIP3) and non-canonical (BIRC3, TRAF3, MAP3K14) NF-κB pathways and were mutually exclusive, pointing to multiple molecular mechanisms targeting NF-κB in SMZL (Fig.1B). By combining mutations and CNAs, NF-κB was affected in 36/101 (36%) SMZL. The TRAF3/MAP3K14-TRAF2/BIRC3 regulatory complex of the non-canonical NF-κB pathway was targeted in 25/101 (25%) SMZL. BIRC3 was affected in 11/101 (11%) SMZL by inactivating mutations (3 frameshift and 2 non-sense), missense mutations (n=1), and gene deletions (n=5). All inactivating mutations were somatically acquired, were predicted to generate aberrant transcripts carrying premature stop codons, and caused elimination or truncation of the C-terminal RING domain, whose E3 ubiquitin ligase activity is essential for MAP3K14 proteosomal degradation by BIRC3 (Fig. 1C). TRAF3 was affected in 10/101 (10%) SMZL by inactivating mutations (2 frameshift and 1 non-sense), missense mutations (n=1) and deletions (n=7). Inactivating mutations were predicted to generate aberrant transcripts carrying premature stop codons and causing elimination or truncation of the C-terminal MATH domain that provides the docking site for MAP3K14, and is required for MAP3K14 recruitment to BIRC3 degradation. MAP3K14 was affected by gain (n=7) or missense mutations (n=1) in 8/101 (8%) SMZL. The missense mutation was somatically acquired and mapped near the kinase domain, suggesting a potential role in MAP3K14 downstream signaling activation. SMZL primary cells harboring BIRC3, TRAF3 or MAP3K14 mutations displayed constitutive NF-κB activation, including MAP3K14 accumulation and active NFKB2 processing by Western blot, and/or nuclear NF-κB localization by immunohistochemistry (Fig. 1D). In addition to non-canonical signaling, also the canonical NF-κB pathway was targeted in SMZL (15/101, 15%) by genetic lesions of TNFAIP3 and IKBKB, which encode for negative and positive regulators of NF-κB signaling, respectively. TNFAIP3 was affected in 13/101 (13%) SMZL by inactivating mutations (6 frameshift) and deletions (n=9), including a focal loss pointing to TNFAIP3 as the specific target of deletions. IKBKB was mutated in 3/101 (3%) SMZL carrying a recurrent, somatically acquired missense mutation (K171E) targeting the IKBKB kinase domain near the activation loop. This mutation leads to an amino acid substitution within a site that is conserved both intra- and inter-species, and is predicted as possibly damaging according to PolyPhen-2. The occurrence of NF-κB molecular lesions in SMZL was observed independent of HCV infection (p=.402), IGHV gene mutation status (p=.065), IGHV1-2 gene usage (p=.761), stereotyped VH CDR3 (p=.969), 7q deletion (p=.621), or TP53 disruption (p=.638), suggesting that genetic deregulation of NF-κB is not restricted to specific SMZL clinico-molecular subgroups. The identification of NF-κB mutations in SMZL elucidates the molecular basis of a fraction of these lymphomas, and expands the genetic mechanisms activating NF-κB in B-cell neoplasia. Beside its pathogenetic relevance, the identification of NF-κB as a pathway recurrently involved in SMZL provides the rationale for targeted anti-NF-κB therapeutic approaches in this lymphoma.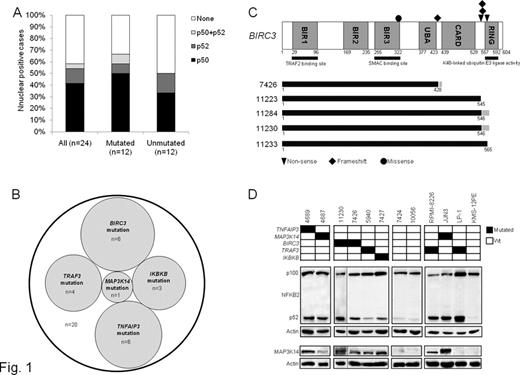

Disclosures:
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal