

Abstract
Clonality can be established by a lack of mosaicism in a female because of random inactivation of either the maternal or paternal X chromosome early in embryogenesis. The methylation status of CpG sites close to the trinucleotide repeats in exon 1 of the human androgen receptor (AR) X chromosome gene assay (HUMARA) has been used to determine clonality. This HUMARA at times indicated clonal hematopoiesis in healthy elderly women, thus precluding its applicability. We used a clonality assay based on quantitative expression of polymorphic X chromosome genes (qTCA) and found no evidence of clonal hematopoiesis in healthy nonanemic elderly persons. We found instances of discordance between HUMARA results and those obtained by pyrosequencing and qTCA methods, as well as by directly quantifying AR gene expression. To determine the basis of this discrepancy we examined the methylation pattern of the AR locus subject to HUMARA. Notably, we found the extent of DNA methylation to be highly variable at the AR gene in granulocytes of persons with discordant results and also in erythroid burst-forming unit colonies but not in those with clonal hematopoiesis. These data provide the molecular basis of incomplete correlation with the pattern of DNA methylation of this X chromosome AR gene locus.
Introduction
Determination of clonality on the basis of assays of inactivation of genes encoded on the X chromosome has provided important insights into the origins of neoplastic diseases, helped to decipher the hierarchy of hematopoiesis, and provided a mechanism for differentiating between inherited and acquired disorders, for distinguishing between clonal and reactive acquired hematologic abnormalities and for showing cell selection in female carriers of X-linked inherited processes.1 The characteristics that make a clonality assay informative and widely applicable include the following. (1) The gene or genes that are being assayed must undergo X-inactivation such that only one isoform is faithfully expressed in each individual somatic cell. (2) The gene or genes of interest need to be sufficiently polymorphic so as to be useful for analysis in a high proportion of the population. (3) The assay should be quantitative because skewing of X-inactivation is a normal biologic process determined by the portion of cells with either an active paternal or maternal X chromosome, a process that occurs randomly during embryogenesis wherein only few cells contribute to the tissues studied. (4) The gene or genes being interrogated should be widely expressed and the assay should be sufficiently robust so that accurate quantitative determinations can be made with a variety of tissues. The discovery of trinucleotide repeats in exon 1 of the androgen receptor (AR) gene (> 1000 bp downstream from AR gene promoter and transcription initiation site) and assessing the DNA methylation status of adjacent CpG sites provided a convenient, fast, and widely used human androgen receptor assay (HUMARA) for measuring clonality2 and appeared to fulfill these criteria, although it does not directly quantify gene or protein expression.3
Oligoclonal hematopoiesis based on skewed AR methylation was reported in healthy elderly women; thus, it was suggested that clonality assays based on X chromosome inactivation are not applicable for the elderly.4-6 However, the methylation status of loci on the inactive X chromosome is heterogeneous, with some loci on inactive X chromosome hypomethylated, whereas others are hypermethylated or had no recognizable methylation differences.7,8 As a result, we have developed an alternate method that differentiates active from inactive X chromosome genes in females, known as transcriptional clonality assays (TCAs).7-10 We have provided evidence that oligoclonal or clonal hematopoiesis was not present in 40 healthy women (all having normal blood counts; age range, 65-92 years; median age, 82 years) when assessed by the quantitative TCA (qTCA), whereas approximately one-third of these women had skewed methylation patterns of the AR locus.11 These results have been challenged by a report of concordance between methylation of the AR locus and X chromosome transcriptional allelic usage.12 However, the healthy persons with skewed methylation of the AR locus were not separately analyzed for skewing of X chromosome allelic usage by qTCA, and it also appeared that the qTCA was not performed as originally described.13
To address these issues, we extended our original experimental design to include studies in young, healthy controls with the use of another well-validated quantitative method, pyrosequencing, to measure expression of polymorphic X chromosome genes; these experiments were performed in a blinded fashion by an investigator at a separate institution (J.J.). The results of the 2 qTCA methods were compared pairwise with results from both the HUMARA method and quantitative expression of AR. Herein, we report a concordance between the 2 qTCA methods, whereas in cases of extreme skewing of X-inactivation as determined by HUMARA in healthy subjects but not in subjects with clonal hematopoiesis, we observed discordance with the results of qTCA and pyrosequencing. These results further validate the accuracy of our qTCA and indicate that extreme skewing of X chromosome inactivation is not a common variation of normal hematopoiesis.
Methods
Study subjects
This study included the following 2 groups of prospectively recruited subjects: (1) young, healthy women (n = 31; mean age, 35 years; median age, 34 years); (2) women with JAK2V617F-positive myeloproliferative neoplasms (6 with polycythemia vera and 1 with essential thrombocythemia); and (3) 3 healthy men. After signing an approved University of Utah Institutional Review Board informed consent in accordance with the Declaration of Helsinki, 10 mL of peripheral blood was obtained by venipuncture. Granulocyte, platelet, and mononuclear cell fractions were isolated according to previously published protocols.14,15
Genotyping of X chromosome single nucleotide exonic polymorphisms
Genomic DNA was isolated from granulocytes with the use of the Puregene DNA purification kit (Gentra). Genotyping of single nucleotide exonic polymorphisms from 5 X chromosome genes all previously shown to be subject to X chromosome inactivation [dbSNP1135363 (C/T) in BTK; dbSNP 9018 (G/A) in FHL1; dbSNP 1141608 (C/T) in IDS; dbSNP 2230037 (C/T) in G6PD; and dbSNP 1126762 (G/T) in MPP1] was performed with TaqMan allele-discrimination assays11 and analyzed with a 7500 Sequence Detection System (Applied Biosystems), as described by the manufacturer. The 3 X chromosome genes (FHL1, IDS, and MPP1) with the greatest frequency of heterozygosity were selected for comparison of expression by both qTCA and pyrosequencing.
Quantitative TCA
Total RNA was isolated from granulocytes and platelets with the use of Tri-Reagent (Molecular Research Center) and used for assessment of allelic usage ratio of the 3 most informative X chromosome polymorphic genes. Aliquots of 1 μg of total RNA were treated with the DNA-free Kit (Ambion) to remove any contaminating DNA. Aliquots of 200 ng of DNA-free RNA were reverse-transcribed with SuperScript VILO for quantitative RT-PCR (Invitrogen). Quantitative allele-specific PCR was performed11 with a Sequence 7500 Detection System (Applied Biosystems), in which an allele-specific primer, containing both a mismatched nucleotide and a locked nucleic acid, is used to enhance discrimination between the polymorphic nucleotides with the use of reaction conditions previously described.11 Aliquots of cDNA from informative subjects (those heterozygous at any of the X chromosome loci) were shipped to MD Anderson Cancer Center for independent qTCA analyses by pyrosequencing in the laboratory of one of the authors (J.J.).
DNA-HUMARA
HUMARA was performed as previously described.11,16 DNA-HUMARA uses a methylation-sensitive nuclease to digest the unmethylated (active) allele. In this assay, allelic ratios were determined by an equation that calculates the amount of PCR product generated by the allele with the longest microsatellite. The relative amount of the shorter of the 2 alleles (allele A) is assigned a value (as percentage) by subtracting the amount of the longer allele (allele B) in percentage from 100%. Therefore, unlike the qTCA, the frequency of each allele is not directly quantified in separate PCR reactions in DNA-HUMARA. PCR products generated from granulocyte DNA were quantified with an ABI PRISM 3130 Automatic Genetic Analyzer (Applied Biosystems), and the ratio of active to inactive X chromosome was determined as described by Bolduc et al.16 Briefly, the direction of methylation skewing was determined on the basis of the quantity of allele B (the allele that harbors the greatest number of CAG repeats). Because of preferential amplification of the smaller allele (A) during PCR, the fraction of HpaII-digested alleles (A and B) was corrected by the fraction of undigested (A′and B′) alleles. These calculations are presented in the following equation:
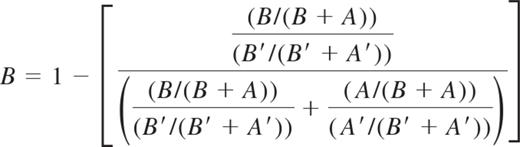
RNA-HUMARA
This RNA-HUMARA is based on AR RNA expression, with polymorphisms determined by the length of the microsatellite cDNA transcripts of exon 1 of AR. In this assay, the PCR cDNA products are subjected to capillary electrophoresis, and the allele expression frequency is determined by quantitating the amount of each of the 2 different size alleles represented in the PCR product. We quantified AR transcripts with the method described by Busque et al.2 Total RNA was isolated from granulocytes with the use of Tri-Reagent (Molecular Research Center) and treated with DNA-free Kit reagents (Ambion) to remove any contaminating DNA. Next, 200 ng of DNA-free RNA were reverse-transcribed with SuperScript VILO in preparation for quantitative RT-PCR (Invitrogen). A mock reverse transcription reaction lacking reverse transcriptase was performed to ensure that no residual DNA contaminated the cDNA preparations. Two microliters of AR cDNA was added to 18 μL of PCR mix and, after amplification, the PCR products were analyzed and quantified with an ABI PRISM 3130 Automatic Genetic Analyzer (Applied Biosystems).
Quantification of SNPs by pyrosequencing analysis
We developed pyrosequencing assays for quantitative detection of exonic single nucleotide polymorphisms (SNPs) in the following X chromosome genes: FHL1, rs9018; IDS, rs1141608; MPP1, rs1126762; AR, rs6153, rs9332969, rs9332970, rs9332971, rs9332972, and rs6154. To decrease the expense and labor, pyrosequencing was not developed for BTK and G6PD exonic polymorphisms because of their lower polymorphic rates among US women.1,17 Granulocyte genomic DNA and cDNA were prepared in Salt Lake City, and aliquots were shipped to MD Anderson Cancer Center. Granulocyte cDNA and genomic DNA (40 ng per reaction) were amplified with gene-specific primers in 2-step seminested PCRs. The second-step PCR was used to label 1 DNA strand with biotin with the use of a universal primer tag biotinylated at the 5′ end.18 Each PCR step was performed in a total volume of 20 μL of 67mM Tris-HCl (pH 9), 16mM ammonium sulfate, 2mM MgCl2, 0.125mM dNTPs, 1 unit of Taq polymerase, and 100 nmol/L PCR primers. A reversible inhibitor of Taq polymerase, TQ21-11 oligonucleotide,19 20 nmol/L, was used in the first-step PCR to achieve a hot start.
The sequences of PCR and pyrosequencing primers are listed in supplemental Table 1 (see the Supplemental Materials link at the top of the article). The following PCR conditions were used: initial denaturation at 95°C for 5 minutes, followed by 40 cycles of denaturation at 94°C for 15 seconds, and annealing/extension at 60°C for 1 minute. We measured the percentage of polymorphic single nucleotides by pyrosequencing with the PSQ HS 96 Pyrosequencing System (QIAGEN) and Pyro Gold CDT Reagents (QIAGEN), as previously described.20
BFU-E analysis
Bisulfite modification of DNA
Genomic DNA from granulocytes, BFU-E colonies, and an induced pluripotent stem cell (iPSC) line (PVb1.11 p19)23 were modified by sodium bisulfite EpiTect Bisulfite kit (QIAGEN) according to the manufacturer's protocol.
Seminested 2-step bisulfite-specific HUMARA exon1 PCR
For the HUMARA exon1 amplification, a seminested 2-step bisulfite PCR approach was used. Two pairs of primers were used for the first PCR reaction (HUMARABis-F-agtgTagttagggTtgggaagggtTtaTTT and HUMARABis-R-ACRCAACCTCTCTCRAAATAACACTCCAAA) and for the seminested reaction (HUMAraBis-NF-gaagggtTtaTTTtYGgTYGTYGtTTAagaTTtaT and HUMARABis-R). PCR reactions were performed in 50-μL reaction that contained 20mM Tris pH 8.4, 50mM KCl, 1.5mM MgCl2, 0.2mM dNTP, 250nM of each primer, 1X Enhancer, and 0.5 U of Taq polymerase (Invitrogen). Thirty-five cycles were performed in MJ research PTC-200 Peltier Thermal Cycler, with the following parameters: 5 minutes at 94°C initial denaturation, and cycling 30 seconds at 94°C, 30 seconds at 60°C for the first PCR reaction, and 58°C for the seminested reaction, and 30 seconds at 72°C. In the second seminested reaction 2 μL of the first PCR product was used.
Methylation detection at control imprinted genes H19 and SNRPN
Two autosomal genes subjected to imprinting were used as a control for our methylation analyses. For the H19 (imprinted maternally expressed transcript [nonprotein coding]) in promoter region amplification, a seminested bisulfite PCR approach was used. Two pairs of primers were used for the first PCR reaction: H19-F, AGGTGTTTTAGTTTTATGGATGATGG, and H19-R, ATAAATATCCTATTCCCAAATAACCCC, and for the seminested H19-FN, TATATGGGTATTTTTGGAGGTTTTT. PCR reactions were performed in 50 μL of reaction containing 20mM Tris pH 8.4, 50mM KCl, 1.5mM MgCl2, 0.2mM dNTP, 250nM of each primer, 1X Enhancer, and 0.5 U of Taq polymerase (Invitrogen). Thirty cycles were performed in MJ research PTC-200 Peltier thermal Cycler with the following parameters: 5 minutes at 94°C initial denaturation, and cycling for 30 seconds at 94°C, 30 seconds of annealing at 60°C, and 30 seconds at 72°C. In the second seminested reaction, 2 μL of the first PCR product was used. The SNRPN (small nuclear ribonucleoprotein polypeptide N) exon 1 amplification was performed as described by Zeschnigk et al.24
Bisulfite sequencing and DNA methylation analysis
After purification of PCR products with the QIAquick gel extraction kit (QIAGEN), individual PCR products were inserted into pCR2.1-TOPO vector (TOPO TA Cloning kit; Invitrogen). Single Escherichia coli colonies positive for recombinant plasmid were picked up, and, after overnight incubation, plasmid DNA was purified with QIAprep Spin Miniprep Kit (QIAGEN) and the PCR insert was sequenced with an ABI PRISM 3130 Automatic Genetic Analyzer (Applied Biosystems).
Sequencing data for CpG methylation were analyzed with the program QUMA (Quantification tool for methylation analysis; Laboratory for Mammalian Epigenetic Studies, Center for Developmental Biology, RIKEN).
Statistical analyses
Shapiro-Wilk tests were applied to determine the normality of the distribution of each analyzed marker for both the HUMARA and qTCA clonality methods. Because the distributions were not statistically significantly nonnormal, Pearson correlation coefficients were calculated to assess the linear relation between the HUMARA and qTCA clonality methods for each analyzed marker. When the distributions were statistically significantly nonnormal, Spearman correlation coefficients (ρ) were calculated. All calculations were performed with SAS Version 9.1 software (SAS Institute).
Results
Comparison of qTCA and quantitative pyrosequencing with DNA-HUMARA and RNA-HUMARA in patients with JAK2V617F+ myeloproliferative disorders
In all informative patients with myeloproliferative neoplasms, platelets and granulocytes were clonal when examined by both qTCAs and were fully concordant with the results of granulocyte HUMARA DNA analysis, as shown in Table 1. Clonality was observed regardless of the JAK2V617F allele burden (data not shown),22 confirming the previous report that the JAK2V617F+ cells represent a subclone of the underlying myeloproliferative neoplasm.22 These data confirm that both qTCA and DNA-HUMARA unambiguously detect clonality in myeloproliferative neoplasms.25 However, when HUMARA-RNA was analyzed in these clonal subjects, the results for patient P4, (Table 1), were not consistent with clonality (assuming clonality is allele ratio of ≥ 75:25).
Results of clonality assays for 7 patients with myeloproliferative disorders
| Sample | Diagnosis | JAK2V617F allele burden (%) | Allelic frequencies (%) of expressed exonic polymorphisms in granulocyte RNA | HUMARA assay* | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| MPP1 G/T (qTCA)† | MPP1 G/T (P)‡ | FHL1 C/T (qTCA)† | FHL1 C/T (P)‡ | IDS C/T (qTCA)† | IDS C/T (P)‡ | Genotype§ | Allele ratios DNA | Allele ratios RNA | |||
| P1 | PV | 97 | 95/5 | 91/9 | 1/99 | 3/97 | 270/281 | 91:9‖ | 100:0 | ||
| P2 | PV | 76 | 100/0 | 97/3 | 4/96 | 11/88 | 270/278 | 10:90 | 0:100 | ||
| P3 | PV | 94 | 92/8 | 97/3 | 270/275 | 0:100 | 3:97 | ||||
| P4 | PV | 14 | 88/12 | 80/20 | 20/80 | ND | 270/272 | 0:100 | 37:63 | ||
| P5 | PV | 72 | 6/94 | 3/97 | 272/285 | 0:100 | 0:100 | ||||
| P6 | ET | 91 | 93/7 | 80/20 | 264/267 | 100:0 | 98:2 | ||||
| P7 | PV | 33 | 2/98 | 4/96 | 10/90 | 4/96 | 264/281 | 0:100 | 0:100 | ||
| Sample | Diagnosis | JAK2V617F allele burden (%) | Allelic frequencies (%) of expressed exonic polymorphisms in granulocyte RNA | HUMARA assay* | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| MPP1 G/T (qTCA)† | MPP1 G/T (P)‡ | FHL1 C/T (qTCA)† | FHL1 C/T (P)‡ | IDS C/T (qTCA)† | IDS C/T (P)‡ | Genotype§ | Allele ratios DNA | Allele ratios RNA | |||
| P1 | PV | 97 | 95/5 | 91/9 | 1/99 | 3/97 | 270/281 | 91:9‖ | 100:0 | ||
| P2 | PV | 76 | 100/0 | 97/3 | 4/96 | 11/88 | 270/278 | 10:90 | 0:100 | ||
| P3 | PV | 94 | 92/8 | 97/3 | 270/275 | 0:100 | 3:97 | ||||
| P4 | PV | 14 | 88/12 | 80/20 | 20/80 | ND | 270/272 | 0:100 | 37:63 | ||
| P5 | PV | 72 | 6/94 | 3/97 | 272/285 | 0:100 | 0:100 | ||||
| P6 | ET | 91 | 93/7 | 80/20 | 264/267 | 100:0 | 98:2 | ||||
| P7 | PV | 33 | 2/98 | 4/96 | 10/90 | 4/96 | 264/281 | 0:100 | 0:100 | ||
ET indicates essential thrombocytosis; P, pyrosequencing; and PV, polycythemia vera.
DNA HUMARA is the clonality assay that uses a methylation sensitive nuclease to digest the unmethylated (active) allele. For DNA and RNA HUMARA assay and principle see “Methods.” The values shown are the mean of duplicate determinations.
Results were based on quantitative transcription-based assay. Values shown are the mean of duplicate determinations.
Results were based on pyrosequencing. Values shown are the mean of duplicate determinations.
Genotype is based on the length of the PCR product that in turn is determined by the length of the exon 1 microsatellite.
The highest value shown is always the putative active (unmethylated) allele.
Comparison of qTCA and pyrosequencing analyses with HUMARA results
Genotyping of exonic SNPs in X chromosome genes MPP1, FL1, and IDS and the AR locus exon 1 microsatellite marker.
Twenty-five of 31 (81%) of the healthy volunteer women were found to be heterozygous in ≥ 1 of the 3 polymorphic X chromosome genes included in this study, whereas 29 of 31 (94%) were heterozygous, based on allelic differences in the number of CAG repeats in the AR locus (Table 2). This rate of heterozygosity with the use of these 2 approaches (informativeness for the clonality analyses) is not significantly different at P = .1297.
Results of clonality assays for 31 healthy young controls
| Sample | Allelic frequencies (%) of expressed exonic polymorphisms in granulocyte RNA | HUMARA assay* | |||||||
|---|---|---|---|---|---|---|---|---|---|
| MPP1 G/T (qTCA)† | MPP1 G/T (P)‡ | FHL1 C/T (qTCA)† | FHL1 C/T (P)‡ | IDS C/T (qTCA)† | IDS C/T (P)‡ | Genotype§ | Allele ratios DNA | Allele ratios RNA | |
| YC1 | 46/54 | 44/56 | 50/50 | 48/52 | 275/283 | 64:36‖ | 55:45 | ||
| YC4 | 30/70 | 37/62 | 70/30 | 62/38 | 275/292 | 33:67 | 67:33 | ||
| YC7 | 37/64 | 21/79 | 275/281 | 45:55 | 64:36 | ||||
| YC9 | 66/34 | 57/43 | 34/66 | 38/62 | 44/56 | 41/56 | 260/270 | 60:40 | 53:47 |
| YC10 | 43/57 | 38/61 | 47/53 | 45/55 | 45/55 | 43/57 | 258/278 | 40:60 | 49:51 |
| YC11 | 42/58 | 49/51 | 55/45 | 43/57 | 267/272 | 47:53 | 58:42 | ||
| YC13 | 41/59 | 31/69 | 267/287 | 27:73 | 47:53 | ||||
| YC14 | 30/70 | 31/69 | 86/14 | 62/38 | 272/278 | 48:52 | 38:62 | ||
| YC15 | 45/65 | 41/58 | 264/275 | 48:52 | 56:44 | ||||
| YC20 | 65/35 | 65/35 | 40/60 | 57/43 | 60/40 | 68/32 | 281/287 | 39:61 | 52:48 |
| YC22 | 27/73 | 23/77 | 43/57 | 39/61 | 266/269 | 33:67 | 39:61 | ||
| YC24 | 45/55 | 44/56 | 258/281 | 46:54 | 79:21 | ||||
| YC25 | 51/49 | 51/49 | 269/289 | 48:52 | 84:16 | ||||
| YC26 | 38/62 | 33/67 | 263/283 | 51:49 | 54:46 | ||||
| YC27 | 77/23 | 76/24 | 78/22 | 75/25 | 272/275 | 68:32 | 56:44 | ||
| YC30 | 48/52 | 49/51 | 266/269 | 49:51 | 50:50 | ||||
| YC31 | 55/45 | 51/49 | 58/42 | 50/50 | 263/272 | 40:60 | 48:52 | ||
| YC12 | 270/281 | 52:48 | 27:73 | ||||||
| YC16 | 278/283 | 69:31 | 56:44 | ||||||
| YC28 | 272/289 | 48:52 | 37:63 | ||||||
| YC6 | 61/39 | 50/50 | 270/270¶ | ||||||
| YC23 | 44/56 | 38/62 | 40/60 | 30/70 | 278/278¶ | ||||
| YC17# | 84/16 | 81/19 | 80/20 | 82/18 | 270/283 | 85:15 | 26:74 | ||
| YC8# | 43/57 | 38/62 | 32/68 | 47/53 | 272/275 | 77:23 | 74:26 | ||
| YC19# | 72/28 | 73/27 | 30/70 | 22/78 | 267/281 | 78:22 | 58:42 | ||
| YC3# | 69/31 | 71/29 | 278/283 | 19:81 | 91:9 | ||||
| YC5# | 42/58 | 54/46 | 51/49 | 56/44 | 275/292 | 10:90 | 36:64 | ||
| YC18# | 68/32 | 63/37 | 267/283 | 16:84 | 0:100 | ||||
| YC2** | 275/278 | 9:91 | 8:92 | ||||||
| YC21** | 272/289 | 21:79 | 0:100 | ||||||
| YC29** | 269/275 | 0:100 | 61:39 | ||||||
| Sample | Allelic frequencies (%) of expressed exonic polymorphisms in granulocyte RNA | HUMARA assay* | |||||||
|---|---|---|---|---|---|---|---|---|---|
| MPP1 G/T (qTCA)† | MPP1 G/T (P)‡ | FHL1 C/T (qTCA)† | FHL1 C/T (P)‡ | IDS C/T (qTCA)† | IDS C/T (P)‡ | Genotype§ | Allele ratios DNA | Allele ratios RNA | |
| YC1 | 46/54 | 44/56 | 50/50 | 48/52 | 275/283 | 64:36‖ | 55:45 | ||
| YC4 | 30/70 | 37/62 | 70/30 | 62/38 | 275/292 | 33:67 | 67:33 | ||
| YC7 | 37/64 | 21/79 | 275/281 | 45:55 | 64:36 | ||||
| YC9 | 66/34 | 57/43 | 34/66 | 38/62 | 44/56 | 41/56 | 260/270 | 60:40 | 53:47 |
| YC10 | 43/57 | 38/61 | 47/53 | 45/55 | 45/55 | 43/57 | 258/278 | 40:60 | 49:51 |
| YC11 | 42/58 | 49/51 | 55/45 | 43/57 | 267/272 | 47:53 | 58:42 | ||
| YC13 | 41/59 | 31/69 | 267/287 | 27:73 | 47:53 | ||||
| YC14 | 30/70 | 31/69 | 86/14 | 62/38 | 272/278 | 48:52 | 38:62 | ||
| YC15 | 45/65 | 41/58 | 264/275 | 48:52 | 56:44 | ||||
| YC20 | 65/35 | 65/35 | 40/60 | 57/43 | 60/40 | 68/32 | 281/287 | 39:61 | 52:48 |
| YC22 | 27/73 | 23/77 | 43/57 | 39/61 | 266/269 | 33:67 | 39:61 | ||
| YC24 | 45/55 | 44/56 | 258/281 | 46:54 | 79:21 | ||||
| YC25 | 51/49 | 51/49 | 269/289 | 48:52 | 84:16 | ||||
| YC26 | 38/62 | 33/67 | 263/283 | 51:49 | 54:46 | ||||
| YC27 | 77/23 | 76/24 | 78/22 | 75/25 | 272/275 | 68:32 | 56:44 | ||
| YC30 | 48/52 | 49/51 | 266/269 | 49:51 | 50:50 | ||||
| YC31 | 55/45 | 51/49 | 58/42 | 50/50 | 263/272 | 40:60 | 48:52 | ||
| YC12 | 270/281 | 52:48 | 27:73 | ||||||
| YC16 | 278/283 | 69:31 | 56:44 | ||||||
| YC28 | 272/289 | 48:52 | 37:63 | ||||||
| YC6 | 61/39 | 50/50 | 270/270¶ | ||||||
| YC23 | 44/56 | 38/62 | 40/60 | 30/70 | 278/278¶ | ||||
| YC17# | 84/16 | 81/19 | 80/20 | 82/18 | 270/283 | 85:15 | 26:74 | ||
| YC8# | 43/57 | 38/62 | 32/68 | 47/53 | 272/275 | 77:23 | 74:26 | ||
| YC19# | 72/28 | 73/27 | 30/70 | 22/78 | 267/281 | 78:22 | 58:42 | ||
| YC3# | 69/31 | 71/29 | 278/283 | 19:81 | 91:9 | ||||
| YC5# | 42/58 | 54/46 | 51/49 | 56/44 | 275/292 | 10:90 | 36:64 | ||
| YC18# | 68/32 | 63/37 | 267/283 | 16:84 | 0:100 | ||||
| YC2** | 275/278 | 9:91 | 8:92 | ||||||
| YC21** | 272/289 | 21:79 | 0:100 | ||||||
| YC29** | 269/275 | 0:100 | 61:39 | ||||||
P indicates pyrosequencing; and YC, young control.
DNA HUMARA is the clonality assay that uses a methylation-sensitive nuclease to digest the unmethylated (active) allele. For DNA- and RNA-HUMARAs and principle, see “Methods.” The values shown are the mean of duplicate determinations.
Results are based on quantitative transcription-based assay. Values shown are the mean of duplicate determinations.
Results are based on pyrosequencing. Values shown are the mean of duplicate determinations.
Genotype is based on the length of the PCR product that in turn is determined by the length of the exon 1 microsatellite.
The highest value shown is always the putative active (unmethylated) allele.
Shown are the 2 non-polymorphic HUMARA samples.
Results of samples that are considered clonal by DNA-HUMARA with ≥ 1 polymorphic genes included in the qTCA/pyrosequencing analysis.
Results of samples that are considered clonal by DNA-HUMARA without a polymorphic gene for analysis by qTCA/pyrosequencing.
Comparison of allelic expression ratios of X chromosome exonic SNPs by qTCA and pyrosequencing.
Both qTCA and quantitative pyrosequencing were performed with cDNA generated from RNA extracted from peripheral blood granulocytes, assaying exonic SNPs in MPP1, IDS, and FHL1 (Table 2). For each individual gene, the same SNP was quantified by both qTCA and pyrosequencing (Table 2). In the case of the qTCAs, each polymorphic allele was quantified in a separate PCR reaction, with any difference in amplification efficiency between allele-specific PCR reactions being taken into account in the equation used to calculate the frequency of expression of each of the 2 polymorphic alleles for each gene.22,26 Although the results shown in Table 2 are the mean of duplicate determination, we validated the accuracy and reproducibility of the assay by randomly selecting samples for analysis in separate PCR reactions (data not shown).
In addition, we compared qTCA results generated using platelet cDNA with results using granulocyte cDNA, and qTCA data in those women heterozygous for > 1 gene using multiple informative gene analyses. In both evaluations (data not shown), we found a high degree of correlation as previously reported.9-11
In pyrosequencing analyses, quantification of expression of polymorphic alleles is based on the heights of the peaks generated on the pyrograms by each allele (supplemental Figure 1). When the results of qTCA were compared with those of pyrosequencing, a good correlation was observed for each of the 3 genes analyzed (Figure 1).

Linear regression analysis. Linear regression analysis of the correlation between allelic expression ratios for qTCA (y-axis) and pyrosequencing (qPyro; x-axis) for 3 markers MPP1 (top left), FHL1 (top right), and IDS (bottom left). Data points were generated according to data in Table 2. For each gene, a statistically significant correlation (P ≤ .0001) was observed when expression based on qTCA was compared with that based on quantitative pyrosequencing.
Linear regression analysis. Linear regression analysis of the correlation between allelic expression ratios for qTCA (y-axis) and pyrosequencing (qPyro; x-axis) for 3 markers MPP1 (top left), FHL1 (top right), and IDS (bottom left). Data points were generated according to data in Table 2. For each gene, a statistically significant correlation (P ≤ .0001) was observed when expression based on qTCA was compared with that based on quantitative pyrosequencing.
Comparison of qTCA with DNA-HUMARA and RNA-HUMARA in the cohort of young, healthy women
Comparison of qTCA with DNA-HUMARA.
Twenty-nine subjects (age range, 22-55 years; mean age, 35 years; median age, 34 years) were found to be heterozygous on the basis of the number of CAG repeats in exon 1 of AR (Figure 2A; Table 2). On the basis of DNA-HUMARA, 9 of 29 of healthy young females (31%) had extreme skewing of granulocyte AR methylation pattern (to allow direct comparison with published HUMARA data defined as an allele ratio of ≥ 75:2512 ; Table 2), a finding similar to that observed in our previous study in which 9 of 26 elderly healthy volunteers (35%) were found to have an extremely skewed ratio of exon 1 differentially methylated microsatellite polymorphic repeats of AR gene.11 Of the 9 subjects with extreme skewing of the AR methylation pattern, 3 were homozygous (noninformative) for any of the 3 genes used in the qTCA/quantitative pyrosequencing assay (Table 2) and 6 were informative (Table 2). One subject (YC17) also had extreme skewing of X-inactivation in both the qTCA and the quantitative pyrosequencing assay; she is healthy, with normal blood counts and no historical evidence of being a carrier for any X chromosome–linked disorders subject to selection.1 The results of these experiments show that, unlike the case for the DNA-HUMARA, extreme skewing of X-inactivation is far less frequent in normal young women when qTCA or quantitative pyrosequencing are used, yet it was in our small study still detected in 1 of 29 samples. In this person the mechanism of extreme skewing may be random, selection for any of X chromosome alleles exerting the different proliferative/survival effect1 at hematopoietic progeny, or any other known or not yet delineated factor(s) effecting X chromosome inactivation.

Comparison of allelic expression ratios generated by qTCA, DNA-HUMARA, and RNA-HUMARA. (A) qTCA compared with DNA-HUMARA. The values for qTCA are the mean of all polymorphic genes for each informative subject shown in Table 2. DNA-HUMARA results are from Table 2. The Pearson correlation coefficient was used to calculate the linear relation (indicated by the line) between qTCA and DNA-HUMARA, with the shaded area above and below the line indicating the confidence intervals. (B) qTCA compared with RNA-HUMARA. The values for qTCA are the mean of all polymorphic genes for each informative subject shown in Table 2. RNA-HUMARA results are from Table 2. The Pearson correlation coefficient was used to calculate the linear relation (line) between qTCA and DNA-HUMARA, with the shaded areas above and below the line indicating the confidence intervals. (C) DNA-HUMARA compared with RNA-HUMARA. The values for DNA-HUMARA and RNA-HUMARA are from Table 2. The Pearson correlation coefficient was used to calculate the linear relation (line) between DNA-HUMARA and RNA-HUMARA, with the shaded area above and below the line indicating the confidence intervals.
Comparison of allelic expression ratios generated by qTCA, DNA-HUMARA, and RNA-HUMARA. (A) qTCA compared with DNA-HUMARA. The values for qTCA are the mean of all polymorphic genes for each informative subject shown in Table 2. DNA-HUMARA results are from Table 2. The Pearson correlation coefficient was used to calculate the linear relation (indicated by the line) between qTCA and DNA-HUMARA, with the shaded area above and below the line indicating the confidence intervals. (B) qTCA compared with RNA-HUMARA. The values for qTCA are the mean of all polymorphic genes for each informative subject shown in Table 2. RNA-HUMARA results are from Table 2. The Pearson correlation coefficient was used to calculate the linear relation (line) between qTCA and DNA-HUMARA, with the shaded areas above and below the line indicating the confidence intervals. (C) DNA-HUMARA compared with RNA-HUMARA. The values for DNA-HUMARA and RNA-HUMARA are from Table 2. The Pearson correlation coefficient was used to calculate the linear relation (line) between DNA-HUMARA and RNA-HUMARA, with the shaded area above and below the line indicating the confidence intervals.
Comparison of qTCA with DNA-HUMARA and RNA-HUMARA.
To analyze the transcripts of the AR gene, we developed a quantitative pyrosequencing assay for 6 AR exonic SNPs separate from the microsatellite repeats measured by HUMARA (rs6153, rs9332969, rs9332970, rs9332971, rs9332972, and rs6154), but these SNPs were found to be insufficiently polymorphic among subjects in the study cohort to be informative (Figure 2B-C). Therefore, to compare the results generated by DNA-HUMARA with those derived from an expression-based assay that used microsatellite RNA repeats measured by HUMARA, the method of Busque et al was implemented.2 Discordance between methylation analyses and transcriptional analyses of the AR gene was observed in 5 subjects (YC17, YC19, YC3, YC5, and YC29), with extreme skewing determined by DNA-HUMARA but not by RNA-HUMARA (Table 2). In 3 tested samples (YC17, YC3, and YC29), the direction of skewing involved opposite alleles, as detected by DNA-HUMARA compared with RNA-HUMARA (Table 2). These results indicate that the methylation pattern of this particular AR locus does not always equate with silencing of its expression.
A separate analysis of the data in Table 2 also found discordance between results generated by qTCA/pyrosequencing and both DNA-HUMARA and RNA-HUMARA. To analyze these relations, for each informative subject, the mean of the proportion of the dominant allele of the informative polymorphic genes, as determined by qTCA, were compared with those obtained by DNA-HUMARA (Figure 2A) and RNA-HUMARA (Figure 2B). In both cases, the linear relation was found to be statistically insignificant (P ≥ .05), although the relation between qTCA and DNA-HUMARA approached statistical significance (P = .0504; Figure 2A). Moreover, direct comparison of DNA-HUMARA results with those generated by RNA-HUMARA was also shown to be statistically insignificant (Figure 2C). These results support the hypothesis that the pattern of DNA methylation does not faithfully reflect X-inactivation or determine AR allele expression. However, that RNA-HUMARA does not correlate with qTCA results also suggests that silencing of AR expression on the inactive X chromosome may be incomplete in healthy women.
Analysis of clonality in the individual BFU colonies of young healthy women
Individual BFU-E colonies from subject YC17 were analyzed for the clonality by DNA-HUMARA and qPCR. One-hundred sixty BFU-E colonies were analyzed by DNA-HUMARA; Sufficient amount of RNA for qTCA analyses from 97 colonies each indicating a single allelic transcript were obtained; this included 40 colonies assayed for MPP1 (34 colonies expressed G allele and 6 T allele) and 10, 16, 14, and 17 colonies assayed for G6PD, IDS, FHL1, and BTK expression, respectively, for G6PD (8 colonies expressed C allele and 2 T allele), IDS (13 colonies expressed C allele and 3 expressed T allele), FHL1 (12 colonies expressed C allele and 2 expressed T allele), BTK (14 colonies expressed C allele and 3 expressed T allele). In contrast, only from 35 individual colonies we were able to amplify exon 1 of the AR gene for both digested and undigested DNA HUMARA analyses (data not shown).
Patterns of CpG methylation of the human androgen receptor gene HUMARA by bisulfite sequencing in native selected granulocytes and colonies from individual BFU-E
To determine the molecular basis of the discrepancy between qTCA analyses and HUMARA, we assessed the methylation status of CpG loci of the AR DNA segment that was subject to HUMARA with the use of standard bisulfite treatment. Granulocyte DNA from healthy controls whose analyses of X chromosome inactivation by DNA- and RNA-HUMARAs and qTCA were discordant (YC3, YC17, YC18, and YC25) were examined and showed in each instance a heterogeneous pattern of methylation at AR locus, as shown in Figure 3A. In contrast, patients with myeloproliferative neoplasms (PV-3, PV-5, and PV-7) showed more homogenous methylation patterns of the AR locus, as shown in supplemental Figure 2A. As a control, we examined the methylation pattern of granulocyte DNA from healthy male controls (bearing a single active X chromosome) which showed a fully unmethylated AR locus, indicating a lack of bias of the bisulfite analysis procedure (supplemental Figure 2B). The predominance of shorter allele A is evident from our results, which is most probably caused by preferential amplification of this allele, as seen also with DNA-HUMARA. Although DNA-HUMARA has implemented correction in the final calculation of allele ratios, this has not been applied for correction of CpG methylation pattern shown here. Furthermore, the heterogeneous pattern of methylation in this polymorphic exon 1 AR locus was also detected in clonal single-cell progenies in all individual in vitro expanded single erythroid progenitors, as analyzed by BFU-E colonies (Figure 3B) from subject YC17; again, both methylated and unmethylated AR DNA were clearly detected in the same BFU-E colony.

Patterns of CpG methylation of the human androgen receptor gene HUMARA by bisulfite sequencing. (A top) Diagram of the 5′ region of the human androgen receptor gene showing the locations of core promoter region, transcription start site (+1), translation start site (ATG), and methylation-sensitive enzyme sites for HpaII. Specific CpG dinucleotides in exon1 region are highlighted in the sequence of PCR product. (down) The sequencing results of the PCR products of the exon 1 HUMARA gene in native selected granulocytes. Total 20-29 clones were analyzed from healthy controls YC25, YC17, YC3, and YC18, respectively. The white dots represent the unmethylated CpGs, whereas black dots represent the methylated CpGs. Short allele (A) and long allele (B) were determined according to the length of the AR gene exon 1 microsatellite. (B) Patterns of CpG methylation of the human androgen receptor gene HUMARA by bisulfite sequencing in individual BFU-Es of subject YC17. The sequencing results of the PCR products of the exon 1 HUMARA gene. Total 6-10 clones were analyzed for BFU-E 1, BFU-E 2, BFU-E 3, BFU-E 4, BFU-E 5, and BFU-E 6, respectively. The white dots represent the unmethylated CpGs, whereas the black dots represent the methylated CpGs. Short allele (A) and long allele (B) were determined according to the length of the AR gene exon 1 microsatellite.
Patterns of CpG methylation of the human androgen receptor gene HUMARA by bisulfite sequencing. (A top) Diagram of the 5′ region of the human androgen receptor gene showing the locations of core promoter region, transcription start site (+1), translation start site (ATG), and methylation-sensitive enzyme sites for HpaII. Specific CpG dinucleotides in exon1 region are highlighted in the sequence of PCR product. (down) The sequencing results of the PCR products of the exon 1 HUMARA gene in native selected granulocytes. Total 20-29 clones were analyzed from healthy controls YC25, YC17, YC3, and YC18, respectively. The white dots represent the unmethylated CpGs, whereas black dots represent the methylated CpGs. Short allele (A) and long allele (B) were determined according to the length of the AR gene exon 1 microsatellite. (B) Patterns of CpG methylation of the human androgen receptor gene HUMARA by bisulfite sequencing in individual BFU-Es of subject YC17. The sequencing results of the PCR products of the exon 1 HUMARA gene. Total 6-10 clones were analyzed for BFU-E 1, BFU-E 2, BFU-E 3, BFU-E 4, BFU-E 5, and BFU-E 6, respectively. The white dots represent the unmethylated CpGs, whereas the black dots represent the methylated CpGs. Short allele (A) and long allele (B) were determined according to the length of the AR gene exon 1 microsatellite.
Methylation detection at control genes H19 and SNRPN
To further determine the reliability of the bisulfite method, we examined the methylation status of CpGs in 2 autosomal imprinted genes H19 and SNRPN.24,27 Both genes were expressed in the iPSC line, but only SNRPN was expressed in granulocytes (data not shown). Aligned with expectations, the H19 gene in iPSC line showed a uniformly and completely methylated pattern on one allele and a lack of methylation on the other allele, in contrast to the results obtained from granulocytes, where this gene is not expressed and where only a methylated allele was found (supplemental Figure 2C). In addition, the SNRPN gene from granulocytes was completely unmethylated on one allele and methylated on the other (supplemental Figure 2C bottom right). Taken together, our controls verify the accuracy of our bisulfite determinations and support our ability to determine the concordance of DNA methylation with gene expression.
Discussion
Detection of clonality on the basis of X chromosome inactivation requires discrimination of the active from the inactive X chromosome. Discovery of a polymorphic tri-nucleotide tandem repeat in exon 1 of AR gene that is subject to differential methylation was exploited to develop the convenient HUMARA clonality assay, with its usefulness confirmed in multiple applications. HUMARA results were generally shown to correlate with X chromosome inactivation, and the method expanded the feasibility of clonality studies because of the high frequency with which microsatellite polymorphisms are observed in the AR locus and the relative ease of obtaining DNA from tissues of interest and their analysis.1,3 However, in some cases, the clonality data obtained by methylation-based HUMARAs were difficult to interpret.28,29 Alternative methods were developed to investigate clonality on the basis of quantitative analysis of the expression of genes subject to X-inactivation.7,9 Previously, we reported a quantitative TCA that was based on thermostable ligase detection.7-9 This method, however, was cumbersome technically and required a large amount of radioisotope, making it impractical for large-scale clinical studies. We have described the qTCA method to measure allele-specific expression of 5 X chromosome genes on the basis of quantification of exonic SNPs.11,28,30 Although based on allele-specific PCR, qTCA is more robust than the standard method because it uses primers that include both a mismatched nucleotide and an artificial locked nucleic acid in the design.22 These modifications enhance the stability of extension products, thereby improving both the specificity and discrimination power of the assay and has greater sensitivity than a widely used pyrosequencing method, wherein the analyzed products < 5% of total are difficult to detect.22
With the use of this qTCA, we revisited the reported phenomenon of extreme skewing of X-inactivation ratios in myeloid blood cells of aging women.11 We studied 40 healthy, elderly women with the use of our qTCA and did not find X chromosome monoallelic expression or extreme skewing. Yet when we analyzed the same subjects with the use of DNA-HUMARA, we found, in agreement with the studies of others,4-6 a monoallelic AR methylation pattern in approximately one-third of the subjects. These data have been challenged by Busque et al,12 who reported concordance between DNA-HUMARA and 2 expression-based clonality assays, including one patterned after our qTCA. However, we noted differences in their study design compared with ours. Specifically, the healthy persons with skewed methylation of the AR locus were not separately analyzed for skewing of X chromosome allelic usage by one or both qTCA methods; their qTCA method differed in their primer design, only one polymorphic gene (IDS) was analyzed, and single cell expanded hematopoietic colonies were not studied. In our studies, we used 5 X chromosome conservative SNPs of genes shown to be subject to complete X chromosome inactivation, and 2 separate blood cell types, both granulocytes and platelets, were analyzed with concordant results.11 To resolve this controversy, we recruited subjects with clonal hematopoiesis, and the results of DNA-HUMARA and qTCA were concordant.
We then analyzed 31 healthy female volunteers (age range, 22-55 years; mean age, 35 years; median age, 34 years). First, the aliquots of blinded samples were assayed by qTCA method, as well as by a well-established quantitative pyrosequencing method for the detection of SNP based on the detection of pyrophosphate release during nucleotide incorporation, which was performed in another institution by an independent investigator (J.J.). In this report, we confirm the reproducibility and accuracy of qTCA in the evaluation of the X chromosome allelic expression (Figure 1). However, in some instances, a lack of concordance between these 2 separate qTCA and DNA-HUMARA results (Figure 2A) was again noted. Analogous to previously obtained results for the healthy elderly women, we found some younger healthy women with extreme skewing and apparent monoallelic methylation patterns of the AR locus (Table 2) that were discordant with results of allele expression ratios obtained by qTCA (Figure 2B). Moreover, we also observed a lack of concordance of the AR gene allelic expression ratios when the locus DNA methylation was compared with its expression in some studied healthy women (YC17, YC3, YC29; Table 1; Figure 2C). The basis of this disparity is speculative, but a plausible explanation is that methylation of the CpG sites in exon 1 of the AR gene is distant from its promoter and may not always correlate with gene silencing. Furthermore, its transcription-start site (also > 1 kb downstream from polymorphic microsatelite exon 1 region) is not located in a CpG island.29,31 DNA methylation is an important component of epigenetic regulation of gene expression; however, total methylation content does not always correlate with gene expression because the methyl content of the active X chromosome has been reported to be twice as high as that for its inactive counterpart.32 Rather, X-specific methylation of CpG islands determines the extent of gene expression from the gene linked to the island, and, as noted above in this paragraph, HUMARA does not assess methylation of AR CpG promoter islands. Thus, we suggest that discordance occurs when the methylation status of the exon 1 region does not extend to the CpG island and promoter region. Thus, the promoter and exon 1 region may be a location where DNA methylation dynamics take place in the cells of healthy women, where full or partial demethylation may contract and expand, resulting in a heterogeneous pattern; a dynamic that may be compromised or lost in clones derived from myeloproliferative disorders (see next paragraph).
Discordance between results of qTCA compared with those of RNA-HUMARA (Figure 2B) implies incomplete or inconsistent silencing of AR expression. This interpretation is based on studies that used a mouse/human somatic cell hybrid system, showing that the genes analyzed in the qTCA are completely and consistently silenced on the inactive X chromosome.33 Although the basis of the proposed incomplete/inconsistent silencing of the AR gene is uncertain, leaky expression from the inactive X chromosome has been reported for a number of genes.13 Incomplete silencing may result from failure of the initial spreading of inactivation along the chromosome, lack of an X-inactivation maintenance step in progeny, or both. Paradoxically, methylation of some loci have been reported to be associated with gene activation,32 and genomic DNA sequence has been implicated in the control of human X chromosome inactivation,34-36 with the presence of short repetitive motifs contributing to the escape process. However, on the basis of results achieved from individual BFU-E colonies, we confirmed that all 5 X chromosome genes MPP1, FL1, IDS, G6PD, and BTK had their inactive X chromosome allele completely silenced at progenies of a single cell expanded in vitro for 14 days in BFU-E assays. In contrast to the observations in the control cohort, concordance between the results of qTCA and DNA-HUMARA were observed for patients with JAK2V617F+ myeloproliferative neoplasms (Table 1). Thus, the processes that confound interpretation of DNA-HUMARA when applied to myeloid cells from healthy subjects appear absent in clonal myeloid myeloproliferative neoplasms.
To determine the basis of the discrepancy in the HUMARA and qTCA results observed in certain healthy subjects, we examined the methylation pattern of the AR locus used for the HUMARA in patient granulocytes with the use of bisulfite sequencing. Persons with discordant HUMARA and qTCA had a heterogeneous pattern of methylation at the polymorphic exon 1 AR locus (Figure 3A). Further, we also found the heterogeneous methylation patterns at this AR locus in the individual in vitro–expanded single erythroid progenitors to BFU-E colonies (Figure 3B). In contrast, patients with myeloproliferative neoplasms who represent clonal hematopoiesis show more homogenous methylation patterns of the AR locus, as shown in supplemental Figure 2A. Presently, the basis of this difference of methylation patterns between normal and clonal tissues remains obscure. However, the technical veracity of our data were confirmed when the same bisulfite sequencing was applied to single X chromosome male DNA and monoallelically expressed imprinted H19 and SNRPN autosomal genes.
Definition of clonality based on qTCA
The exact timing of molecular mechanism of X chromosome inactivation is not exactly known. It is generally agreed that this first recognized epigenetic process is mediated by XIST (X-inactive–specific transcript) RNA. Some work suggests that the XIST RNA accumulates on 1 of the 2 X chromosomes in the blastocyst around the 8-cell stage and transcriptionally silences this X chromosome.37 Other work suggests that there is a considerable diversity in the timing of this process in placental mammals, and in humans the XIST RNA accumulates only in the postblastocyst stage.38
However, our previous studies,10,11 and those of others,39 that used the distribution of ranges of X chromosome transcript usage ratios in healthy women10,11 and work with normal human T cells/mouse cell hybrids39 have shown that X-inactivation is a stochastic process that occurs at about the 8-cell stage of embryogenesis. On the basis of these observations, we developed a model to predict the probability of skewing of X-inactivation in healthy subjects (Figure 4). According to this model, the probability of a healthy subject having an allele expression ratio of 100:0 is ∼ 0.4%, the probability of having a ratio of 87.5:12.5 is ∼ 3%, and the probability of having a ratio of 75:25 is ∼ 11%. On the basis of this information, we use a ratio of ≥ 80:20 to define clonality, accepting that ∼ 5% of healthy women will have skewing of X-inactivation sufficient to be classified as clonal hematopoiesis. This assumption is consistent with observations from the cohort described herein, because 1 (YC17) of 25 (4%) of the informative subjects had an allele ratio of ≥ 80:20 (Table 2). The number of false-positive controls could be reduced by increasing the ratio, but doing so would probably eliminate some true-positive samples, as shown by the results in Table 1 (patients P4 and P6). In this situation, clonal myeloid cells may be contaminated by nonclonal T lymphocytes, or the granulocytes may be composed of variable proportions of monoclonal and polyclonal cells. A corollary argument is that a clone may expand insufficiently to shift the allele ratio into the clonal category, resulting in a false-negative interpretation. Our clonality assay system has additional controls designed to reduce false-positive and false-negative results, because concordance among all informative polymorphic genes must be observed (as many as 5 genes may be polymorphic in a test subject11 ) and both granulocyte and platelet cDNA is assayed.11 Moreover, as noted in “Comparison of qTCA with DNA-HUMARA and RNA-HUMARA in the cohort of young healthy women” of “Results,” qTCA directly quantifies both polymorphic alleles, in contrast to HUMARA in which one allele is quantified directly and the quantity of the other allele is derived.

Distribution of expected counts given 8 independent equiprobable trials, each with a probability of 0.5. This figure show the probability that somatic tissue in an adult will have a particular allele ratio, assuming stochastic inactivation of either the maternal or paternal X chromosome at the 8-cell stage of embryogenesis and no positive or negative selection over time based on X chromosome gene expression. The x-axis indicates the number of either maternal or paternal alleles that are active. For example, 8 could indicate that all of the maternal alleles are active. In that case, 8 paternal alleles would be inactive, and the allele expression ration would be 100:0. Similarly, 7 would indicate that 7 maternal and 1 paternal gene are active. In that case, the allele ration would be 87.5:12.5. The probability of each ratio is as follows: 0:8, 0.004; 1:7, 0.031; 2:6, 0.109; 3:5, 0.219; and 4:4, 0.273.
Distribution of expected counts given 8 independent equiprobable trials, each with a probability of 0.5. This figure show the probability that somatic tissue in an adult will have a particular allele ratio, assuming stochastic inactivation of either the maternal or paternal X chromosome at the 8-cell stage of embryogenesis and no positive or negative selection over time based on X chromosome gene expression. The x-axis indicates the number of either maternal or paternal alleles that are active. For example, 8 could indicate that all of the maternal alleles are active. In that case, 8 paternal alleles would be inactive, and the allele expression ration would be 100:0. Similarly, 7 would indicate that 7 maternal and 1 paternal gene are active. In that case, the allele ration would be 87.5:12.5. The probability of each ratio is as follows: 0:8, 0.004; 1:7, 0.031; 2:6, 0.109; 3:5, 0.219; and 4:4, 0.273.
The observations reported herein further validate the use of qTCA for analysis of clonal hematopoiesis and provide the molecular basis of the incomplete correlation of HUMARA and qTCA results seen in some persons because of the heterogeneous pattern of DNA methylation at this particular AR gene locus.
This article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgment
The authors thank Dr Alexander Gimelbrant for helpful suggestions and critical review. The authors dedicate this paper to the memory of the late Ernest Beutler who pioneered the idea of transcriptional clonality assay and who encouraged them to elucidate discrepancies between HUMARA and transcriptional clonality assay, which are finally presented in this paper.
This work was supported by the National Cancer Institute (grant 1P01CA108671-O1A2), the Myeloproliferative Disorders (MPD) Consortium (project #1; J.T.P. principal investigator), Howard Hughes Medical Institute (B.C.), and Palacky University (internal grant LF_2011_011).
National Institutes of Health
Authorship
Contribution: S.I.S. designed and performed the research, analyzed data, and wrote the paper; L.P. helped to design the methylation experiments and assisted with performance of methylation analyses and critically reviewed the paper; J.J. contributed to the design of the research, performed pyrosequencing experiments, analyzed data, and wrote the paper; N.A. analyzed data, accrued study subjects and obtained their consent and wrote the paper; S.H. and B.R.C. designed the analyses of methylation pattern of AR locus and provided reagents and assisted with the methylation techniques used; A.W. performed statistical analysis and reviewed the manuscript; K.H. recruited study subjects and obtained their consent and reviewed the manuscript; C.J.P. contributed to design of the research, analyzed data, and critically reviewed the paper; B.R.C. played crucial roles in suggesting the use of imprinted gene controls and critically reviewed the data and paper; and J.T.P. conceived the project, designed the study, analyzed data, and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Josef T. Prchal, Hematology Division, University of Utah, 30 North 1900 East, SOM 5C402, Salt Lake City, UT 84132; e-mail: josef.prchal@hsc.utah.edu.
