

Abstract
During thrombotic or hemostatic episodes, platelets bind collagen and release ADP and thromboxane A2, recruiting additional platelets to a growing deposit that distorts the flow field. Prediction of clotting function under hemodynamic conditions for a patient's platelet phenotype remains a challenge. A platelet signaling phenotype was obtained for 3 healthy donors using pairwise agonist scanning, in which calcium dye–loaded platelets were exposed to pairwise combinations of ADP, U46619, and convulxin to activate the P2Y1/P2Y12, TP, and GPVI receptors, respectively, with and without the prostacyclin receptor agonist iloprost. A neural network model was trained on each donor's pairwise agonist scanning experiment and then embedded into a multiscale Monte Carlo simulation of donor-specific platelet deposition under flow. The simulations were compared directly with microfluidic experiments of whole blood flowing over collagen at 200 and 1000/s wall shear rate. The simulations predicted the ranked order of drug sensitivity for indomethacin, aspirin, MRS-2179 (a P2Y1 inhibitor), and iloprost. Consistent with measurement and simulation, one donor displayed larger clots and another presented with indomethacin resistance (revealing a novel heterozygote TP-V241G mutation). In silico representations of a subject's platelet phenotype allowed prediction of blood function under flow, essential for identifying patient-specific risks, drug responses, and novel genotypes.
Introduction
During a clotting event, platelets respond to combinatorial stimuli, including collagen, adenosine diphosphate (ADP), thromboxane A2 (TXA2), thrombin, epinephrine, and serotonin, as well as endothelial-derived inhibitors such as nitric oxide and prostacyclin (PGI2). Excessive platelet buildup at the site of cardiovascular disease and plaque rupture causes more than 1 million heart attacks and strokes in the United States each year. Excessive thrombus formation after plaque rupture remains difficult to predict and can be linked to hyperactive platelet function.1,2 Therefore, low-dose aspirin to inhibit platelet cyclooxygenase-1 (COX-1) and clopidogrel to inhibit platelet P2Y12 signaling are used widely in patients with cardiovascular risks, although patient response to these drugs can vary.
Interindividual variations in platelet reactivity, even in the healthy population, have been associated with several factors, including female sex, fibrinogen level, ethnicity, inherited variations, and polymorphisms.3,4 Similarly, platelet dysfunction or antiplatelet therapy can be associated with bleeding risks.5-7 Furthermore, the function of blood is highly dependent on hemodynamic forces; examples include shear-induced platelet activation at > 5000/s shear rate,8,9 requirement of VWF in arterial thrombosis,10-12 shear effects on VWF structure/function and GPIb-VWF A1 domain–bonding dynamics,13-17 RBC-dependent platelet migration toward the wall,18,19 and convection-enhanced mass transfer to and from local zones of clotting or bleeding.20,21 Defining, quantifying, and linking a patient's unique platelet phenotype or coagulation phenotype under hemodynamic conditions to clinical risk remains a diagnostic challenge.
In the presence of multiple activating stimuli, platelet signal transduction results in calcium mobilization, leading to inside-out signaling, including integrin activation, dense granule release of ADP, ATP, serotonin, epinephrine, α-granule release of VWF and thrombospondin, phospholipase A2/COX-1–dependent TXA2 synthesis, and shape change (Figure 1A). In a previous study, we established an efficient PAS method to measure this combinatorial signaling by testing pairs of agonists at low, medium, and high concentrations.22 Neural network (NN) models could therefore be trained that interpolated between the tested doses and extrapolated to predict responses to ternary stimulation, sequential exposures, and exposures of up to 6 agonists.22 The NN model of calcium regulation (Figure 1B) is therefore ideal for use in a newly developed multiscale model (Figure 1C-D) in which platelets convect through boundary layers of ADP and TXA2 while depositing on collagen or on other platelets, where they are exposed to high concentrations of stimulating molecules.
![Figure 1. Multiscale model of combinatorial platelet activation and thrombus formation under flow. Platelet agonists (blue) used individually or in pairs to activate GPVI- or G-protein–coupled receptors (the thromboxane receptor TP, purinergic receptors P2Y1 and P2Y12, and the prostacyclin receptor, IP) result in modulation of intracellular calcium (green) from intracellular stores distal of phospholipase C (PLC) activation or from store operated calcium entry via Stim1-Orai1 activation. Inhibitors (red) such as acetylsalicylic acid (ASA) or indomethacin inhibit COX-1. Autocrine pathways include release of TXA2 and ADP (A). A 2-layer, 8-node/4-node NN with feedback is trained with 74 measured calcium traces to predict [Ca]i for each patient-specific platelet PAS (B). The multiscale simulation of platelet deposition under flow requires simultaneous solution of the instantaneous velocity field over a complex and evolving platelet boundary Ω(t) by LB, concentration fields of ADP and TXA2 by FEM, individual intracellular platelet state ([Ca]i) and release reactions (R) for ADP and TXA2 by NN, and all platelet positions and adhesion/detachment by LKMC (C-D).](/view-large/figure/7277477/zh89991292000001.jpeg)
Multiscale model of combinatorial platelet activation and thrombus formation under flow. Platelet agonists (blue) used individually or in pairs to activate GPVI- or G-protein–coupled receptors (the thromboxane receptor TP, purinergic receptors P2Y1 and P2Y12, and the prostacyclin receptor, IP) result in modulation of intracellular calcium (green) from intracellular stores distal of phospholipase C (PLC) activation or from store operated calcium entry via Stim1-Orai1 activation. Inhibitors (red) such as acetylsalicylic acid (ASA) or indomethacin inhibit COX-1. Autocrine pathways include release of TXA2 and ADP (A). A 2-layer, 8-node/4-node NN with feedback is trained with 74 measured calcium traces to predict [Ca]i for each patient-specific platelet PAS (B). The multiscale simulation of platelet deposition under flow requires simultaneous solution of the instantaneous velocity field over a complex and evolving platelet boundary Ω(t) by LB, concentration fields of ADP and TXA2 by FEM, individual intracellular platelet state ([Ca]i) and release reactions (R) for ADP and TXA2 by NN, and all platelet positions and adhesion/detachment by LKMC (C-D).
Multiscale model of combinatorial platelet activation and thrombus formation under flow. Platelet agonists (blue) used individually or in pairs to activate GPVI- or G-protein–coupled receptors (the thromboxane receptor TP, purinergic receptors P2Y1 and P2Y12, and the prostacyclin receptor, IP) result in modulation of intracellular calcium (green) from intracellular stores distal of phospholipase C (PLC) activation or from store operated calcium entry via Stim1-Orai1 activation. Inhibitors (red) such as acetylsalicylic acid (ASA) or indomethacin inhibit COX-1. Autocrine pathways include release of TXA2 and ADP (A). A 2-layer, 8-node/4-node NN with feedback is trained with 74 measured calcium traces to predict [Ca]i for each patient-specific platelet PAS (B). The multiscale simulation of platelet deposition under flow requires simultaneous solution of the instantaneous velocity field over a complex and evolving platelet boundary Ω(t) by LB, concentration fields of ADP and TXA2 by FEM, individual intracellular platelet state ([Ca]i) and release reactions (R) for ADP and TXA2 by NN, and all platelet positions and adhesion/detachment by LKMC (C-D).
To develop an integrated view of blood function under flow in response to multiple stimuli encountered during thrombosis and hemostasis, in the present study, we used PAS to measure calcium mobilization in platelets obtained from 3 subjects. In addition, whole blood from each of these 3 subjects was perfused over fibrillar collagen using microfluidic devices in which the blood was pretreated with inhibitors of COX (indomethacin or aspirin), P2Y1 (MRS-2179), or with a PGI2 receptor (IP receptor) activator (iloprost). NN models of platelet calcium signaling were then embedded in a multiscale model that simulated platelet deposition to collagen. The multiscale model accounted for changes in blood flow as the platelet deposit grew into the channel, the motion of platelets within the blood flow, and the release of soluble platelet agonists (ADP and TXA2). This allowed for multiscale prediction of donor-specific clotting events under flow and pharmacologic modulation, a fundamental step in describing how a heart attack would progress based on a patient's unique platelet phenotype.
Methods
Blood collection
Blood was drawn from healthy male volunteers (who self-reported to be free of medication or alcohol consumption in the prior week) into H-D-Phe-Pro-Arg-chloromethylketone (100μM PPACK; Calbiochem) to inhibit thrombin production, all at normal extracellular calcium concentration. Donors provided informed consent in accordance with University of Pennsylvania institutional review board approval and the Declaration of Helsinki.
PAS of platelet calcium signaling
Platelet calcium mobilization in response to agonist stimulation was measured in a high-throughput assay, as described previously.22 Diluted PPACK-treated platelet-rich plasma (12% by volume) was treated with indomethacin (28μM) to eliminate autocrine signaling via thromboxane and loaded with Fluo-4NW dye (Invitrogen) for 30 minutes before activation in a 384-well plate assay with all pairwise mixtures of ADP, U46619, and convulxin using a FlexStation III (Molecular Devices) at final concentrations of 0, 0.1, 1, and 10 × EC50 levels (EC50 for ADP, 1μM; for U46619, 1μM; and for convulxin, 5nM). Each combination of agonists was tested in the presence or absence of iloprost (1μM). All wells contained normal extracellular calcium. NN training was also carried out as described previously.22 The NN training set consisted of the concentrations of the 4 molecular inputs (ADP, U46619, CVX, and iloprost) and the resulting calcium transients for the 74 conditions. A unique NN was trained for each donor (see supplemental Methods, available on the Blood Web site; see the Supplemental Materials link at the top of the online article).
Microfluidic phenotyping of platelet
The microfluidic devices were fabricated in poly(dimethylsiloxane) as described previously23 with 8 parallel channels (250 μm wide by 60 μm high) that converged to a common outlet. The channels ran perpendicularly over a 250-μm-long stripe of patterned equine fibrillar collagen type 1 (Chrono-par; Chrono-log), allowing 8 separate platelet deposition events per device to be imaged simultaneously by epifluorescence microscopy (magnification, 4×; 620 nm EX/700 nm EM) every 15 seconds (ORCA-ER CCD camera; Hamamatsu).
Before blood perfusion, channels were blocked with 0.5% BSA in HEPES-buffered saline (20mM HEPES and 160mM NaCl, pH 7.5) for 30 minutes. Blood was treated with vehicle (0.01% final concentration of DMSO) or various concentrations of indomethacin (Tocris Bioscience) or acetylsalicylic acid (aspirin; Sigma-Aldrich) 40 minutes before perfusion or with iloprost or MRS-2179 (Tocris Bioscience) for 5 minutes before perfusion. Alexa Fluor 647–conjugated anti-CD41 (clone PM6/248; AbD Serotec) for 3 minutes before perfusion. Samples were perfused at a wall shear rate of 200/s (2 μL/min per channel; PHD2000 Harvard Syringe pump) for 8 minutes.
Sequencing of the TP gene
DNA was isolated from whole blood using the FlexiGene DNA kit following the manufacturer's instructions (QIAGEN) and subjected to PCR amplification using primers against TP exons 2, 3, and 4 with sequences kindly provided by Andrew Mumford (Bristol Heart Institute, University of Bristol, Bristol Royal Infirmary, Bristol, United Kingdom).24 PCR products were either sequenced directly or subcloned for sequencing by the University of Pennsylvania DNA-sequencing facility.
Calcium measurements in HEK cells expressing wild-type TP or TP-V241G
The wild-type human TXA2 receptor cDNA (TBXA2R clone ID TXA2R0000) was obtained from the University of Missouri cDNA Resource Center. The cDNA construct is cloned into pcDNA3.1+ (Invitrogen) at EcoR1 (5′) and XhoI (3′) for expression under a CMV promoter. The variant V241G TXA2R was generated from this wild-type template by site-directed mutagenesis using a QuikChange II XL Site-Directed Mutagenesis Kit (Agilent Technologies) following the manufacturer's instructions. The mutated clone was sequenced and verified in the University of Pennsylvania DNA-sequencing facility. Equivalent transient expression after lipofection of plasmid for wild-type TP and TP-V241G was verified by flow cytometry using anti–human TP Ab (Cayman Chemical) at 21%-22% transfection. Lipofected HEK-293T (4 μg of plasmid with 10 μL of lipofectamine 2000 per well of a 6-well plate) were cultured for 1 day and then reseeded in 96-well plates with clear bottoms (COSTAR 3603) and cultured 2 days before the calcium measurements. The cells were washed once using HEPES-buffered saline and incubated with the fluorescent Ca2+ indicator Fluo-4NW (Invitrogen) at room temperature for 30 minutes. Fluorescence using Molecular Devices Flex Station III after stimulation with U46619 was normalized to the fluorescence before stimulation (F/Fo) and then background subtracted by the signal from untransfected HEK.
Multiscale simulation of platelet deposition under flow
The multiscale model consisted of 4 submodels: lattice kinetic Monte Carlo (LKMC), NN, finite element method (FEM), and lattice Boltzmann (LB). The LKMC submodel tracked individual platelet motion in the fluid and platelet binding/unbinding to the surface. LKMC is a stochastic method that solves for the motion of particles within flow and can incorporate cellular binding rates. The main input into a LKMC model is the rate for all events, in this case, motion and binding events. The NN submodel provided the concentration of intracellular calcium given a history of agonist exposure for each platelet. The FEM submodel computed the transient concentration profiles of ADP and TXA2 in the fluid as platelets became activated. The FEM solved the convection-diffusion-reaction equation, which incorporates the effects of diffusion and convection on the transport of soluble species. The LB submodel determined the velocity field of the blood flow due to platelet deposition in the channel.
A uniformly discretized (hLKMC = hLB = 0.5 μm, hFEM = 1 μm, where h is the lattice spacing of each method; supplemental Figure 1), 2D-rectangular domain (500 μm long and 60 μm high) was used to represent one microfluidic channel in the vicinity of the collagen patch. The blood velocity field throughout the domain was solved by LB assuming fully developed parabolic flow at the inlet, no-slip boundary conditions at the walls and deposited platelet aggregates, and constant specified pressure at the exit. Blood was assumed to be Newtonian with viscosity of 3 cP. Platelet events considered within the LKMC simulation include platelet diffusion (with rate ΓD = Dplatelet / hLKMC2) and convection (with rate ΓC = v / hLKMC) where Dplatelet is the effective platelet diffusivity because of Brownian and RBC motion25 and v is the local fluid velocity magnitude.26,27 RBC motion creates a nonuniform radial platelet distribution in which the platelet concentration is highest near the walls. This effect was modeled as a platelet radial drift velocity28 that was superimposed on the velocity field obtained from the LB simulation. The resulting LKMC simulation creates a physiologically consistent 3-fold wall excess of platelets.
To calculate platelet activation-dependent rates of attachment and detachment to and from collagen or already deposited platelets, the internal activation state ξ of the ith platelet at time t was defined as the integral calcium concentration above the basal level, ξi (t) = ∫ ([Ca]i − [Ca]0) dt. [Ca]0 was set to 100nM, and the time-dependent calcium concentration was computed using the donor-specific NN, which accounts for exposure to collagen, ADP, TXA2, and iloprost along the platelet trajectory. The integrated calcium concentration, ξ(t), for each platelet was then used to define a time-dependent extent of inside-out signaling, F(ξ), for α2β1 and α2bβ3 integrin activation, as follows:
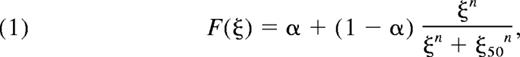
where n controls the sharpness of the response, and ξ50 is the critical value for 50% activation. In the present study, the values of n = 0.75 and ξ50 = 9μM-sec were used for both α2β1 and α2bβ3 integrin activation. The quantity α represents the basal level of integrin activation and was set to 0.001 in all simulations so that 0.001 ≤ F < 1.
The overall rate of attachment of a platelet to collagen is defined by
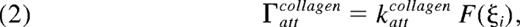
where kattcollagen is the attachment rate of a fully activated platelet (F = 1) and accounts for receptor and ligand copy numbers, single bond kinetics, and the effects of VWF. Similarly, the rate of attachment between 2 platelets via fibrinogen, Γattfibrinogen, depends on the activation states of both platelets. To model the 2-body interaction, the rate of binding between platelets i and j is given by the geometric mean of integrin activation
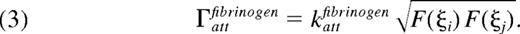
The detachment rate of platelets from collagen, Γdetcollagen is modeled using the Bell exponential29 to describe force-dependent breakage of receptor-ligand bonds,
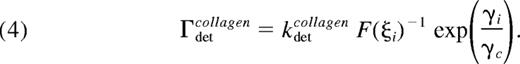
where F (ξi)−1 is a metric of the number of bonds that must be broken, γi is the local shear rate around platelet i, and γc is the characteristic shear rate required to initiate accelerated bond breakage. Similarly, the detachment rate between 2 bound platelets is given by
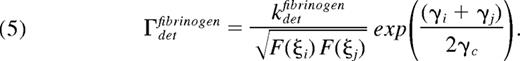
The concentration fields, Cj(x,y,t), (j = ADP and TXA2) were determined by FEM solution of the convection-diffusion-reaction equation,
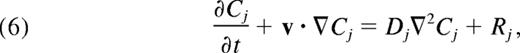
where Dj is the Brownian diffusion coefficient of ADP or TXA2 (augmentation effects for small solutes in blood flow are relatively small30 ). Rj is the volumetric rate of release or generation of ADP or TXA2, and v is the velocity field (obtained from LB). The platelet release rate of soluble species ADP and TXA2 depends on the internal activation state, ξ. We assume that each platelet only releases ADP and TXA2 if its integral calcium, ξi (t) is larger than the critical threshold, ξ50 = 9μM-sec. The time at which a platelet reaches ξ50 is denoted as trelease. For t ≤ trelease, the rate of release of ADP or TXA2 is modeled by an exponential decay of the form
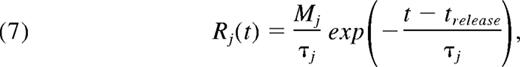
where Mj is the total amount of releasable ADP or TXA2 in a platelet and τj is the characteristic time constant of release. Once activated, each platelet releases 1 × 10−8 nmol of ADP with a time constant of 5 seconds31,32 and 4 × 10−10 nmol of TXA2 with a time constant of 100 seconds.33 All parameters are defined in supplemental Table 1 and were held constant across all donors and drug conditions. To simulate individual donors, a donor-specific NN was embedded in the multiscale model. To simulate the effect of drug treatment, signals were either inhibited or stimulated at the NN level. COX-1 inhibition was modeled by inhibiting TXA2 signaling. P2Y1 inhibition was modeled by inhibiting ADP signaling. Iloprost was a direct input to the NN (see supplemental Methods)
Results
PAS and platelet calcium phenotyping
For each of 3 donors, 74 calcium responses were measured in quadruplicate on a 384-well plate (Figure 2A). The trained NN models predicted these calcium responses accurately within 5% at all times (Figure 2A and supplemental Figure 2). The measured synergies for 63 conditions, containing 2 or 3 inputs (agonists with or without iloprost), provide a “synergy vector” that is a measurement of a subject's phenotype (Figure 2B). We found previously that the synergy vector is a unique and repeatable fingerprint of the subject.22 The NN training resulted in accurate prediction of this measured synergy vector for each donor (Figure 2B-C), with the synergy scores highly correlated (R = 0.9543) between experiment and simulation (Figure 2C).

PAS for platelet calcium mobilization for 3 separate donors. For Donors 1-3, calcium traces were measured in the presence of low, medium, and high doses of ADP, U46619, and convulxin (CVX) in the presence or absence of iloprost and simulated by NN (A). The normalized synergy parameters (−1 ≤ S ≤ 1) for all measured pairs (± iloprost) was calculated for each donor as well as for the donor-specific NN-simulated calcium responses under identical conditions (B). Measured and NN-simulated synergy values were highly correlated, indicating successful training of the NN models (C).
PAS for platelet calcium mobilization for 3 separate donors. For Donors 1-3, calcium traces were measured in the presence of low, medium, and high doses of ADP, U46619, and convulxin (CVX) in the presence or absence of iloprost and simulated by NN (A). The normalized synergy parameters (−1 ≤ S ≤ 1) for all measured pairs (± iloprost) was calculated for each donor as well as for the donor-specific NN-simulated calcium responses under identical conditions (B). Measured and NN-simulated synergy values were highly correlated, indicating successful training of the NN models (C).
Multiscale simulation of platelet function under flow
Multiscale simulations predicted the density of platelets adherent to the surface, platelet activation states, and the spatiotemporal dynamics of ADP and TXA2 release, morphology of the growing aggregate, and the distribution of shear along the solid-fluid boundary (shown in Figure 3 for Donor 1; see supplemental Video 1). Initially, platelets only adhered to the collagen surface until the adherent platelets became sufficiently activated to release ADP and TXA2. The released ADP and TXA2 formed a boundary layer extending up to 10 μm from the platelet deposit, which was sufficient to recruit platelets in the near-wall region. Within the boundary layer, concentrations of up to 10μM ADP and 0.1μM TXA2 were observed. Concentrations of ADP and TXA2 and the boundary layer thickness were consistent with earlier continuum models that imposed a thrombus growth rate and geometry.34 TXA2 concentrations were found to be subphysiologic (< 0.0067μM or < 0.1 × EC50) until a sufficient platelet mass accumulated at the surface after approximately 250 seconds. Boundary-layer ADP concentrations were within the effective dynamic range (0.1-10μM or 0.1-10 × EC50) throughout the simulation. The ADP concentration profile had spatial and temporal fluctuations of 2-3 orders in magnitude, whereas the TXA2 concentration profile had less than an order of magnitude of spatial fluctuations and no temporal fluctuations. Spatial fluctuations in the TXA2 profile were created by an inhomogeneous distribution of platelet density along the surface. The strong temporal and spatial fluctuations in the concentration of ADP were predominately driven by the short release time (5 seconds), whereas the longer release time of TXA2 (100 seconds) smoothed fluctuations. Platelets interacting with collagen became the most activated because of the sustained calcium mobilization in collagen signaling, whereas platelets that were stimulated by only ADP and TXA2 had only a transient increase in intracellular calcium. The platelets participating in the initial platelet-collagen interaction experienced a fluid shear close to the initial wall shear rate of 200/s. The shear rate along the solid-fluid boundary became highly heterogeneous during the simulation, with areas of high shear rate (5- to 10-fold increase above 200/s) at the projections into the flow and areas of near zero shear rate in the valleys between platelet aggregates. Therefore, within a single simulation (and the corresponding microfluidic experiments), a large range of shear rates are sampled by platelets, even in the presence of a constant inlet wall shear rate. Low shear recirculation regions of up to 10 μm were formed within the valleys of the platelet deposit. At 500 seconds, the platelet deposit was characterized by platelet clusters approximately 30 μm in size, which is fully consistent with microfluidic measurements of platelet cluster size on collagen at this shear rate.35

Multiscale simulation of patient-specific platelet deposition under flow. Platelet activation (in all subplots: black indicates inactivated and white, fully activated) and deposition at 500 seconds (inlet wall shear rate, 200/s) for Donor 1 in the presence of released ADP (A) and TXA2 (B) in which the local shear rate near the platelet deposit varied markedly from less than 50/s to greater than 1000/s (C). Flow: left to right (streamlines; black lines); surface collagen (250 μm long): red bar. The background color of each subplot represents the state of a single, representative simulation at 500 seconds for the concentrations of ADP (A) and TXA2 (B) and the local fluid shear rate (C). The activation state (ξ) of each platelet and the morphology of the surface is the same across all subplots.
Multiscale simulation of patient-specific platelet deposition under flow. Platelet activation (in all subplots: black indicates inactivated and white, fully activated) and deposition at 500 seconds (inlet wall shear rate, 200/s) for Donor 1 in the presence of released ADP (A) and TXA2 (B) in which the local shear rate near the platelet deposit varied markedly from less than 50/s to greater than 1000/s (C). Flow: left to right (streamlines; black lines); surface collagen (250 μm long): red bar. The background color of each subplot represents the state of a single, representative simulation at 500 seconds for the concentrations of ADP (A) and TXA2 (B) and the local fluid shear rate (C). The activation state (ξ) of each platelet and the morphology of the surface is the same across all subplots.
The multiscale model relies on 21 parameters (supplemental Table 1) held constant in all simulations, the first 14 of which are well-measured or accurately estimated physical and biologic constants (ρblood, μblood, etc) and the remaining 7 parameters (n, ξ50, kattcollagen, kdetcollagen, kattfibrinogen, kdetfibrinogen, and γc) were constrained by the dynamical responses measured for the 4 conditions across 3 donors (Figure 4), allowing a coarse-grained description of receptor-mediated adhesion. Because α2bβ3, P-selectin, and phosphatidylserine exposure (measured with PAC1, anti–P-selectin, and Annexin V, respectively) were uniform across the 3 donors used in this study in response to strong activation with convulxin (supplemental Figure 3), it was reasonable to treat these 7 coarse-grained parameters as uniform across the set of donors.

Comparison of microfluidic experiments and multiscale simulations of platelet deposition. Measured platelet deposition dynamics for 3 donors in the presence of aspirin, indomethacin, MRS 2179, and iloprost (A) and the corresponding donor-specific multiscale simulations with no TXA2 (indomethacin or aspirin), no ADP (MRS 2179), and iloprost treatment (B). Comparison of measured (C) and simulated (D) platelet deposit mass at 500 seconds for Donors 1-3. Shading for time series data in panels A and B shows SD. Experiment: control, n = 24, n = 22, and n = 22 for Donors 1, 2, and 3, respectively; aspirin, n = 8; indomethacin, n = 8; MRS 2719, n = 8; and iloprost, n = 14. Simulation: n = 16 for all conditions and donors. *P < .01 relative to control for each donor.
Comparison of microfluidic experiments and multiscale simulations of platelet deposition. Measured platelet deposition dynamics for 3 donors in the presence of aspirin, indomethacin, MRS 2179, and iloprost (A) and the corresponding donor-specific multiscale simulations with no TXA2 (indomethacin or aspirin), no ADP (MRS 2179), and iloprost treatment (B). Comparison of measured (C) and simulated (D) platelet deposit mass at 500 seconds for Donors 1-3. Shading for time series data in panels A and B shows SD. Experiment: control, n = 24, n = 22, and n = 22 for Donors 1, 2, and 3, respectively; aspirin, n = 8; indomethacin, n = 8; MRS 2719, n = 8; and iloprost, n = 14. Simulation: n = 16 for all conditions and donors. *P < .01 relative to control for each donor.
Multiscale simulation predicts donor-specific platelet deposition under flow
Using microfluidic devices, real-time platelet deposition from PPACK-treated whole blood flowing over fibrillar collagen (250 μm × 250 μm patch) was measured for 500 seconds at inlet wall shear rate of 200/s for 3 separate donors under untreated conditions or in the presence of aspirin, indomethacin, MRS-2179, or iloprost (Figure 4A and supplemental Video 2). In the 2D simulations, the overall extent of platelet deposition was measured by the number of platelets on a patch with surface area of 750 μm2 (a 250 μm × 3 μm slice; Figure 4B-C) over 500 seconds. In the 2D simulations, a total of 100-300 platelets were deposited on the collagen surface at 500 seconds for the control condition (corresponding to 8000-25 000 platelets on a 250 μm × 250 μm area), which compares very well with the maximal deposition measured in the microfluidic experiment (approximately 20 000 platelets at FI = 3000 in Figure 4A). Several platelet-signaling pathways were examined using drug treatments (Figure 1A). For both experiment and simulation, all donors were the most sensitive to iloprost and the least sensitive to removal of TXA2 via indomethacin or aspirin treatment (TXA2 = 0 in the simulation). The effect of removing ADP-induced calcium mobilization via MRS-2179 treatment (ADP = 0 in the simulation) was between that of iloprost and that of removing TXA2. Blood from Donor 1 was observed to produce significantly larger clots at 500 seconds than blood from Donors 2 and 3 under control conditions (P < .001; Figure 4A), a trait exactly predicted in multiscale modeling (Figure 4B,D). In both experiment and simulation, Donors 1 and 2 exhibited significant sensitivity (P < .01) to the removal of TXA2 (indomethacin or aspirin in the experiment, TXA2 = 0 in the simulation) at 500 seconds, whereas Donor 3 was insensitive to indomethacin or aspirin. For Donors 1 and 2, the effect of removing TXA2 via COX-1 inhibition occurred after approximately 250 seconds in the experiment, which corresponded well to the time at which physiologically active levels of TXA2 were predicted to occur in the boundary layer. In both simulation and experiment, the effect of ADP removal or iloprost treatment occurred after 100 seconds, the time at which propagation of the platelet aggregate away from the clot surface began. The SD of the simulation (because of stochasticity in the LKMC method) was comparable to that observed with the microfluidic chamber replicates.
To test the predictive accuracy of the simulation under hemodynamic conditions of arterial thrombosis, we conducted experiments and simulations with an initial arterial wall shear rate of 1000/s. A total of 48 clotting events (over 3 separate days) were conducted at either 200/s or 1000/s for Donors 2 and 3 (Donor 1 refused continued participation in the 3-day study). The multiscale model predicted a significant decrease (P < .0001, n = 24) in platelet deposition, which was also observed in the microfluidic experiment (supplemental Figure 4). In the simulation, shear rates range from the initial wall shear rate of 1000/s to more than 3000/s at the peaks of the thrombus that perturbed into the flow field.
Donor 3 platelet phenotype reveals a heterozygotic mutation in the TP receptor
In stark contrast to Donors 1 and 2, Donor 3 was insensitive under flow conditions to the COX-1 inhibitors aspirin and indomethacin (Figure 4A,C), a trait predicted exactly in the multiscale modeling (Figure 4D) and fully consistent with an insensitivity to the thromboxane mimetic U46619 (Figure 5A). Platelet aggregation in response to 2μM U46619 was also abnormal for Donor 3 relative to a healthy donor (Figure 5B). After sequencing the 4 exons of the TXA2R gene of Donor 3 that were amplified using the PCR primers of Mumford et al,24 a heterozygous T → G mutation was found at position 11 921 in Exon 2 (National Center for Biotechnology Information reference number NG_013363.1; Figure 5C, supplemental Figure 5, and supplemental Methods), resulting in the identification of a novel heterozygote TP-V241G mutation in Donor 3. Because Gq signaling through ADP was normal in Donor 3 (supplemental Figure 6) and the TP receptor level in Donor 3 was also normal relative to Donors 1 and 2 (supplemental Figure 7), we conclude that the V241G mutation was the cause of abnormal coupling of TPα to Gq and impaired calcium mobilization. Using transient expression of wild-type human TXA2R (TP) or mutated TXA2R (TP-V241G) in lipofected HEK cells (approximately 21% transfection efficiency and equivalent expression for both plasmids; supplemental Figure 8 and supplemental Methods), we detected an impaired calcium response to U46619 in Flou-4NW–loaded HEK expressing the mutated TP receptor relative to responses observed in cells transfected with the wild-type TP receptor (Figure 5D-E).

Linking phenotype to genotype for Donor 3. Unlike Donors 1 and 2, Donor 3 displayed almost no calcium response (time courses obtained from PAS experiments in Figure 2) to increasing doses of U46619 (A). Platelets from Donor 3 were unresponsive to 2μM U46619 in a platelet-rich plasma aggregation assay relative to a healthy donor (B). A heterozygote T → G mutation in the TP receptor was found in Donor 3, resulting in the V241G mutation (C). Transient expression of TP-V241G in HEK cells resulted in cells that were unresponsive to U46619 at conditions that activated calcium mobilization when wild-type TP was expressed in HEK cells (D-E). Peak wild-type TP response to 0.5 or 1nM U46619 was 2.1-fold greater (P < .001; n = 8 replicates averaged in each trace with ± 21% error) than sham stimulation, whereas the peak TP-V241G response was not significantly different from sham stimulation.
Linking phenotype to genotype for Donor 3. Unlike Donors 1 and 2, Donor 3 displayed almost no calcium response (time courses obtained from PAS experiments in Figure 2) to increasing doses of U46619 (A). Platelets from Donor 3 were unresponsive to 2μM U46619 in a platelet-rich plasma aggregation assay relative to a healthy donor (B). A heterozygote T → G mutation in the TP receptor was found in Donor 3, resulting in the V241G mutation (C). Transient expression of TP-V241G in HEK cells resulted in cells that were unresponsive to U46619 at conditions that activated calcium mobilization when wild-type TP was expressed in HEK cells (D-E). Peak wild-type TP response to 0.5 or 1nM U46619 was 2.1-fold greater (P < .001; n = 8 replicates averaged in each trace with ± 21% error) than sham stimulation, whereas the peak TP-V241G response was not significantly different from sham stimulation.
Discussion
A central goal in blood systems biology is to elucidate the regulatory complexity of cellular signaling across a large ensemble of interacting cells responding to numerous spatiotemporal stimuli in the presence of pharmacologic mediators, ideally in a patient-specific and disease-relevant context (ie containing hemodynamics). Because platelet activation is defined by calcium mobilization in this model, it was expected that drug treatment through IP activation, COX1 inhibition, and P2Y1 antagonism in the model would lead to decreased aggregation. However, the multiscale simulation accurately predicted the ranking of pharmacologic sensitivity of blood under flow although the only “knowledge” of drug treatment was through the NN. The effect of COX1 inhibition in Donors 1 and 2 was observed in the microfluidic assay after ∼ 200 seconds of perfusion. This onset of action was also apparent in the simulation and is consistent with aggregometry results showing action of COX1 inhibitors at the later stages after primary aggregation.36 Prediction of individual aggregate sizes corresponded well with experimental observations and such predictions would not be possible with continuum models. In addition, Donor 1 produced significantly larger platelet masses in the control condition than the other donors (P < .001). The difference between donors was captured from the high-dimensional calcium data (concentrations of 3 agonists and 1 inhibitor) predicted by the NN model. Iloprost treatment, however, resulted in the same level of reduced deposition in all 3 donors in both simulation and experiment. Thus, the model is robust to changes in platelet signaling as captured in the NN for each unique donor and in response to multiple drug treatments. Furthermore, PAS and microfluidic assay for platelet phenotyping resulted in identification of a new TP-V241G mutation in Donor 3, the functional impact of which was quantitatively captured in an in silico model of blood from Donor 3 (Figure 4D).
The main mechanism of P2Y12 inhibition is through an increase in the levels of cAMP.37 We investigated the effect of an increase in cAMP by stimulating the IP receptor via iloprost in this study. cAMP levels have been shown to strongly correlate with platelet activation regardless of whether the increase of cAMP was because of P2Y12 inhibition or IP receptor stimulation.38 Our results indicate that cAMP elevation is more effective than COX-1 or P2Y1 inhibition under these conditions.
The multiscale and patient-specific modeling presented here extends earlier modeling efforts of platelet function and/or coagulation20,21,39-42 by including a highly robust description of intracellular platelet signaling through GPVI, P2Y1, TP, and IP. To our knowledge, our NN/multiscale model is the first such approach to make donor-specific predictions of platelet function under flow in the presence of both released ADP and thromboxane and pharmacologic modulators of clinical relevance. However, the model reported here does not account for thrombin production (and all measurements were obtained in 100μM PPACK-treated whole blood). This simplification enables the input of NN phenotyping, consideration of multiple platelet therapies, and direct quantitative comparison to experimental results from microfluidic systems. A continuum coagulation model similar to the Leiderman-Fogelson model21 can be adapted to a discrete platelet model as in Xu et al42 although a computational price is paid for the addition of approximately 50 coupled partial and ordinary differential equations (compared with 2 partial differential equations here). The simulations in Figure 4 required 192 simulations (3 donors × 4 conditions × 16 repeats) each requiring up to 8 hours on Lonestar at the Texas Advanced Computational Center) with a significant computational burden associated with FEM calculation of ADP and TXA2 concentration fields. Whereas it is not intractable to include the effects of thrombin production, the scope of such investigation is quite computationally challenging and would still require a validated description of patient-specific platelet function, the focus of the present study. Under flow conditions, the generation of thrombin by fibrillar collagen surfaces containing tissue factor has a moderate enhancing effect (approximately 30%) on platelet deposition, but a very large effect on fibrin deposition.43 Additional donor-specific measurements of receptor levels, fibrinogen and VWF levels, ADP and TXA2 release, for example, would allow for further tailoring of these parameters for in silico models of blood. Recognizing that kdetcollagen also coarse grains VWF function, we expect this kinetic parameter to be a function of shear rate above approximately 1500/s. Unfortunately, full adhesive dynamic simulations44 for 5 minutes of flow with hundreds of adhering and activating platelets is not computationally feasible, perhaps requiring > 104 hours of CPU per simulation condition, thus the need for the coarse-grained approach.
The TP mRNA is subject to alternative splicing, however, only the TPα isoform is found in platelets. The residue Val241 in the third intracellular loop of the receptor near the inner membrane is highly conserved in the TXA2R gene of humans, mice, pigs, chimpanzees, gibbons, cows, and Xenopus. The V241G mutation is a newly described point mutation that, like the other known TP point mutations of R60L45 and D304N,24 displays normal TP expression levels but poor calcium mobilization and aggregation at 1-2μM U46619. R60L and D304N are associated with a mild bleeding tendency; however, Donor 3 has no self-reported bleeding abnormality. Mutations in TP can result in aspirin resistance, but such mutations are fairly rare, despite the unexpected discovery of a TP mutation phenotype by PAS with only 3 donors in this study. In more than 20 healthy donors studied by PAS to date, only Donor 3 was found to be unresponsive to U46619.
Developing tools to define platelet variations between patients and the relationship of platelet phenotype to prothrombotic or bleeding traits will have significant impact in stratifying patients according to risk. The multiscale model used in the present study also makes feasible patient-specific prediction of platelet deposition and drug response in more complex in vivo geometries such as stenosis, aneurysms, stented vessels, valves, bifurcations, or vessel rupture (for prediction of bleeding risks) or in geometries encountered in mechanical biomedical devices.
There is an Inside Blood commentary on this article in this issue.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the Texas Advanced Computing Center at The University of Texas (Austin, TX) for providing computing resources.
This work was supported by the National Institutes of Health (R01 HL-103419 to S.L.D.).
National Institutes of Health
Authorship
Contribution: M.H.F. developed all of the multiscale simulations; T.C. conducted the microfluidic studies and aggregometry; M.S.C. conducted the PAS and NN training; H.J. and S.Z. conducted the experiments shown in Figure 5C through E; D.J. conducted the flow cytometry; L.F.B. advised on platelet signal transduction; T.S. advised on multiscale simulation code development; and S.L.D. directed the study and wrote the manuscript with contributions from all authors.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Scott L. Diamond, Institute for Medicine and Engineering, Department of Chemical and Biomolecular Engineering, 1024 Vagelos Research Laboratories, University of Pennsylvania, Philadelphia, PA 19104; e-mail: sld@seas.upenn.edu.
