

Abstract
Abstract 1033
Chronic, nonmalignant, noninfectious lymphadenopathy associated with splenomegaly and elevated circulating TCRα β+, CD4-/CD8- Double Negative T (DNT) cells are the hallmarks of the apoptosis disorder called autoimmune lymphpoproliferative syndrome (ALPS). ALPS is most frequently associated with defects in the FAS gene affecting the extrinsic pathway of apoptosis. Patients with ALPS-like syndromes caused by somatic mutations in NRAS and KRAS affecting the intrinsic (mitochondrial) pathway of apoptosis are recently recognized and currently classified separately as ALPS-related apoptosis disorders. Reduced peripheral blood lymphocyte apoptosis after withdrawal of IL-2 is a key feature of their diagnosis. They present with autoimmune phenomena, massive splenomegaly, modest lymphadenopathy, and normal or only marginally elevated DNT cells. Concomitant abnormalities of the myeloid compartment, with an absolute monocytosis with or without concurrent leukocytosis and neutrophilia, mimic juvenile or chronic myelomonocytic leukemia in otherwise asymptomatic younger patients. These patients are classified as RAS-associated autoimmune leukoproliferative disorder (RALD) (Oliveira et al, Blood 2010 and Niemela et al, Blood 2011).
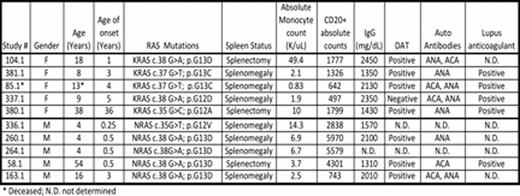
Here we summarize the clinical features in our cohort of 10 patients with RALD. They are currently 4–54 years old (M:F ratio 5:5) and all are alive except one patient who died at age 13. Their median age of disease onset was 2 years; while 50% presented with symptoms at or before age of 1year; one patient (380.1) presented with symptoms as an adult (age 36) with hypersplenism and leukocytosis. All presented with significantly enlarged spleens and 3/10 have undergone splenectomy. All 5 patients with NRAS mutation are male in our cohort, whereas all 5 patients with KRAS mutation are female and three of them presented with ecchymotic periarthritis. As a group these patients have an absolute monocytosis (median 1.7 K/uL, range 0.8–49.7 K/uL), increased circulating B lymphocytes (median 743, range 497–5970) and elevated serum IgG (median 1960 mg/dL, range 1310–2450 mg/dL). Monocytes appear to express unique activation markers in the two patients studied to date. Monocytes from patients 380.1 and 381.1 showed atypical expression of CD56 and 40% of them from patient 381.1 were atypical with an immunophenotype consistent with dendritic cells (non-classical monocytes) that expressed CD16, bright CD45, bright HLA-DR, dim CD14, and dim CD36.
Bone marrow biopsies performed on a subset of patients show hypercellular marrow with monocytosis, granulocytic hyperplasia, and no increase in blasts. Atypical megakaryocytes and myeloid maturation asynchrony have been noted in some, without overt dysplasia. Autoantibodies noted in these patients include DAT, lupus anticoagulant, ACA IgG, ACA IgM, and ANA. Importantly cytokine (IL-2) withdrawal assay showed diminished apoptosis in each of these patients while GM-CSF hypersensitivity assay was positive in the 3/5 patients tested. Only one patient in our cohort (Patient 58.1) with an NRAS mutation has developed B cell lymphoma at age 44 and is alive 10 years after receiving chemotherapy. RALD should be considered in patients with unexplained splenomegaly, autoimmune phenomena and persistent monocytosis. Diligent clinical and laboratory assessments and long-term follow up will help us understand the natural history of this rare leukoproliferative disorder and distinguish it from myelodysplastic conditions like JMML and CMML.
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal