

Key Points
T-cell clones identified in the GI tract of patients with steroid-refractory acute GI GVHD expand in the blood with disease progression.
The T-cell repertoire in the GI tract of steroid-refractory patients at the time of diagnosis is more similar than for responsive patients.
Abstract
Steroid refractory gastrointestinal (GI) acute graft-versus-host disease (aGVHD) is a major cause of mortality in hematopoietic stem cell transplantation (HCT) without immune markers to establish a diagnosis or guide therapy. We found that T-cell receptor β (TCRβ) complementarity-determining region 3 repertoire sequencing reveals patterns that could eventually serve as a disease biomarker of T-cell alloreactivity in aGVHD. We identified T-cell clones in GI biopsies in a heterogeneous group of 15 allogeneic HCT patients with GI aGVHD symptoms. Seven steroid-refractory aGVHD patients showed a more conserved TCRβ clonal structure between different biopsy sites in the GI tract than 8 primary therapy–responsive patients. Tracking GI clones identified longitudinally at endoscopy in the blood also revealed an increased clonal expansion in patients with steroid-refractory disease. Immune repertoire sequencing-based methods could enable a novel personalized way to guide diagnosis and therapy in diseases where T-cell activity is a major determinant.
Introduction
Hematopoietic cell transplantation (HCT) is an effective therapy for a broad range of hematologic malignancies.1-3 The benefits of HCT are in large part related to the development of effective immune responses against the underlying malignancy. However, immune reactivity against the recipient in the form of acute graft-versus-host disease (aGVHD) is a leading cause of morbidity and mortality that limits the use of HCT. aGVHD typically manifests within 100 days of transplant, during which donor T cells attack host skin, gut, or liver in a process fueled by inflammatory signals from host antigen-presenting cells and tissues.2,4 For the 30% to 45% of patients that develop aGVHD, just more than half develop gastrointestinal (GI) tract manifestations, and these aGVHD patients with significant GI involvement account for most of the mortality.1,3
Symptoms of GI aGVHD include nausea, anorexia, diarrhea, abdominal pain, and hemorrhage.5 Severity of these symptoms is a poor predictor of survival, because they also frequently occur in the posttransplant setting secondary to conditioning regimen toxicity, infection, or medications. Endoscopic biopsy confirmation of aGVHD is standard, but histologic grade at diagnosis does not reliably predict disease severity or survival and cannot be used to guide therapy.6
Front-line therapy for GI aGVHD involves increased immunosuppression using corticosteroid and other medications. This initial therapy effectively treats 24% to 41% of patients.6 There is no consensus on treatment of steroid-refractory (SR) GI aGVHD, a subgroup with high mortality. A fundamental obstacle to identifying and treating aGVHD, and thus improving HCT outcomes, is a lack of objective disease biomarkers to guide therapy and standardize patients in studies. Some groups have proposed panels of blood biomarkers of tissue damage to predict aGVHD treatment outcomes.7,8 Alhough promising, these approaches do not monitor alloreactive T cells known to be causally related to disease activity. A minimally invasive, reliable blood measure of T-cell alloreactivity could significantly aid in the management of GI aGVHD.9
Despite decades of research, the identity and activity of the alloreactive donor T cells that cause aGVHD have remained inaccessible to clinical practitioners, largely due to the highly personalized and complex repertoire of T-cell receptor (TCR) hypervariable regions that arises when unique donor T cells are transplanted into recipients with unique histocompatibility antigens.9 Several groups have found an association between oligoclonal T-cell populations and poor outcomes in HCT recipients.10-12 Quantitative studies of patient-specific T-cell repertoires after HCT can now be scaled, due to the use of next-generation sequencing to enumerate the full repertoire of recombined TCR complementarity-determining region 3 (CDR3) repertoire sequences.13-16
We used repertoire sequencing to identify dominant personal T-cell clones in the GI tracts of patients with aGVHD at the time of diagnosis. Here we report the first analysis of the clonal TCRβ repertoire in patients with GI GVHD and find that the T-cell repertoire was more similar at different sites in the GI tracts of patients with severe treatment refractory GI GVHD compared with patients with mild, treatment-responsive aGVHD or with aGVHD-like symptoms without an identified cause. Importantly, each patient had a unique repertoire with little overlap among patients. Last, longitudinal tracking of GI-identified clones in peripheral blood at time of diagnosis and at 30 days after diagnosis revealed statistically significant clonal expansion in patients with SR disease vs primary-responsive (PR) patients.
Methods
Patients and aGVHD classification
Between July 2011 and July 2012, 15 patients provided informed consent in accordance with the Declaration of Helsinki and were enrolled in a research and biobanking protocol approved by the Stanford University Institutional Review Board. Patients were clinically scored for aGVHD using the Beardsman criteria by treating attending physician.17 Patients with known cytomegalovirus (CMV) colitis were excluded.
Sample collection and DNA extraction
For identified patients undergoing HCT with suspected GI aGVHD, we requested that our gastroenterology colleagues collect 1 to 2 upper and 1 to 2 lower GI samples in areas of disease involvement. If no areas showed involvement, samples were to be collected in the right and left colon and the duodenum and stomach. GI biopsy specimens were placed in saline at 4°C for 30 to 120 minutes before removal from saline and freezing at −80°C.
TCRβ repertoire sequencing
Genomic DNA was isolated using standard methods (Qiagen). TCRβ repertoire sequencing was performed using GigaMune Rep-Seqmolecular kits and ClonoByte repertoire analysis software (GigaGen). Briefly, 1.6 µg genomic DNA was amplified by polymerase chain reaction (PCR) with a set of 45 primers targeting the T cell receptor V beta genes paired with 13 primers targeting the T cell receptor J beta genes. This set of 58 primers amplifies the CDR3 region of TCRβ and also introduces universal priming sites to the amplicons. A second round of PCR was performed on the resulting amplicons using universal primers. Each sample was indexed with a unique 6-nucleotide tag, allowing demultiplexing of samples after sequencing. Samples were then sequenced on a Genome Analyzer IIx or MiSeq (Illumina).
The analysis of the sequencing results was performed blinded to whether the patient had SR or PR GVHD. The sequencing reads for each sample were analyzed using ClonoByte TCRβ repertoire analysis software (GigaGen). We aligned each sequence to the set of TRBV and TRBJ genes by identifying the conserved cysteine and phenylalanine that form the boundaries of the CDR3. For quality purposes, we discard reads that do not have a uniquely identifiable V gene, are out of frame as defined by the conserved cysteine and phenylalanine, or contain a stop codon or a sequencing error in the form of an uncalled base. All other nucleotide sequences are translated into their amino acid equivalent.
Errors can accrue during the presequencing PCR and during the sequencing process, distorting the real diversity of the TCRβ CDR3 repertoire by creating a tail of low-abundance clones. We removed the background of spurious low-abundance clones: First, we ran a no-template negative control alongside each batch of 12 samples at every stage. In the rare cases where the no-template negative control showed a strong spurious CDR3 signal, we removed those clones from all corresponding samples. Second, for each sample, we removed all reads mapping to clones whose frequency is <0.1% of the most abundant clone's frequency and then normalized the remaining clones’ frequencies to the total number of remaining reads.
Whole-repertoire statistics
The Bhattacharyya coefficient measures the amount of overlap between 2 statistical samples and was applied to unique clonotypes and their frequencies.18 It is calculated as
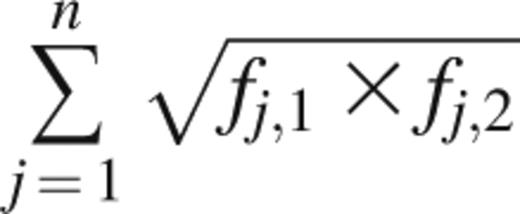
where fj,1 and fj,2 are the frequencies of clonotype j in samples 1 and 2, respectively, and n is the number of unique clonotypes present across samples 1 and 2. A Bhattacharyya coefficient of 1 between 2 Rep-Seq samples would mean that the samples are identical in number, sequence, and frequency of unique clonotypes. A value of 0 would mean that no clonotypes are present in both samples.
The Gini coefficient is a metric of inequality in a frequency distribution.19 It is commonly used to measure economic distributions such as income inequality but has been applied in biology as a measure of oligoclonality of HTLV-1–infected T-cell clones.20 The Gini coefficient is defined as the ratio of the area between the Lorenz curve of the distribution and the line of equality to the area under the line of equality. Thus, a Gini coefficient of 1 corresponds to a completely monoclonal sample containing a single clone, whereas a Gini coefficient of 0 corresponds to a sample with all clones present at equal frequencies. For a Rep-Seq data set containing n unique clonotypes ranked from least frequent (k = 1) to most frequent (k = n), the Gini coefficient can be approximated with the trapezoidal rule as
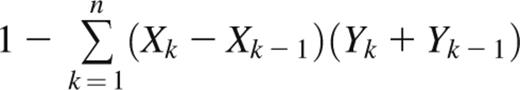
where Xk is the cumulative proportion of unique clonotypes 1,…,k, and Yk is the cumulative frequency of clonotypes 1,…,k.
For patient-level comparisons, statistical evaluation was performed on the mean of any repeated measures (eg, multiple biopsy tissue sites within 1 patient), except as specified. All statistical significance testing was performed using Prism 6 (GraphPad).
Indicator clone index at day 30
The indicator clone index at day 30 (ICI-30) is derived from the average fold change of indicator clone frequencies in peripheral blood over time, weighted by each indicator clone’s frequency in the tissue of discovery. An ICI-30 value of 100 indicates that, on average, indicator clone frequency in blood at day 30 after diagnosis (Dx) remained unchanged from time of diagnosis, whereas a value of 500 indicates that the weighted average fold change was fivefold. Performing repertoire sequencing on a patient’s GI tissue, blood at time of diagnosis, and blood at day 30 post-Dx yields 3 sets of unique CDR3 sequences and their frequencies. From these data, the ICI-30 is calculated as
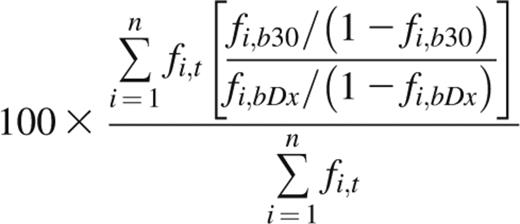
where n is the number of tissue indicator clones in tissue t; and fi,t, fi,bDx, and fi,b30 are the frequencies of clonotype i in tissue, blood at time of diagnosis, and blood at day 30 post-Dx, respectively. Indicator clones that were not detected above background in either blood sample were assigned a fold change of 1 and included in the weighted average. If an indicator clone was detected above background in one blood sample but not the other, we assigned the clone a frequency equal to the background cutoff of the sample from which the clone was absent, ie, 0.1% of the frequency of the most abundant clone.
Results
Patient enrollment and sample collection
We sequenced the TCRβ repertoires of biopsy tissue obtained from a heterogeneous group of 15 consecutive allogeneic HCT patients who underwent endoscopy for suspected GI aGVHD (Table 1). We also obtained blood from each patient at the time of aGVHD diagnosis and at 25 to 35 days after diagnosis (day 30 post-Dx; Figure 1A). We compared 7 HCT patients with aGVHD requiring secondary therapy (SR) and 8 HCT patients who recovered from symptoms seen in aGVHD following primary immunosuppressive therapy (PR). All but 1 of the 15 patients received steroids as primary GVHD therapy (supplemental Table 1 on the Blood website).
Patient characteristics
| Patient no. | Sex | Age | Indication | Conditioning | Donor | Prophylaxis | Stage | CMV | Outcome |
|---|---|---|---|---|---|---|---|---|---|
| 4958 | M | 60 | Plasma cell leukemia | BCNU/Flu/ATG | URD | CSA/MMF | 3 | −/− | Death |
| 4975 | F | 20 | AML | Thio/Flu/ATG | Haplo | Treg | 2 | +/− | Death |
| 5026 | M | 68 | MDS | BCNU/Melph/Flu | URD | CSA/MTX | 2 | −/+ | Death |
| 5072 | F | 46 | Extranodal NKT | BCNU/VP-16/Cy | MRD | FK/PSE | 3 | −/+ | Death |
| 5112 | F | 41 | CML blast crisis | Bu/Cy | URD-1 | FK/MTX | 3 | +/− | Death |
| 5216 | F | 43 | AML | TBI/VP-16/CY | MRD | Treg | 2 | −/+* | Death |
| 5236 | M | 62 | MDS/MPN | BCNU/Melph/Flu | URD | FK/MMF | 2 | −/+† | IR |
| 4986 | M | 55 | MDS | Bu/Cy | URD | FK/MTX | 2 | +/+† | CR |
| 4998 | M | 53 | NHL | BCNU/VP-16/Cy | MRD | FK/PSE | 2 | −/− | CR |
| 5005 | F | 55 | CLL | TLI/ATG | URD-1 | CSA/MMF | 2 | −/+ | No steroids, CR |
| 5182 | F | 60 | Per. T-cell lymphoma | Bu/Cy | URD | FK/MTX | 2 | −/− | CR |
| 5199 | M | 39 | CML blast crisis | TBI/Cy | URD | FK/MTX | 2 | −/+* | CR |
| 5215 | M | 66 | CLL | TLI/ATG | URD | CSA/MMF | 3 | −/− | CR |
| 5261 | M | 53 | ALL | TBI/Cy | URD | FK/MTX | 3 | −/− | CR |
| 5314 | F | 38 | ALL | TBI/VP-16/Cy | URN-1 | FK/MTX | 2 | +/+ | CR |
| Patient no. | Sex | Age | Indication | Conditioning | Donor | Prophylaxis | Stage | CMV | Outcome |
|---|---|---|---|---|---|---|---|---|---|
| 4958 | M | 60 | Plasma cell leukemia | BCNU/Flu/ATG | URD | CSA/MMF | 3 | −/− | Death |
| 4975 | F | 20 | AML | Thio/Flu/ATG | Haplo | Treg | 2 | +/− | Death |
| 5026 | M | 68 | MDS | BCNU/Melph/Flu | URD | CSA/MTX | 2 | −/+ | Death |
| 5072 | F | 46 | Extranodal NKT | BCNU/VP-16/Cy | MRD | FK/PSE | 3 | −/+ | Death |
| 5112 | F | 41 | CML blast crisis | Bu/Cy | URD-1 | FK/MTX | 3 | +/− | Death |
| 5216 | F | 43 | AML | TBI/VP-16/CY | MRD | Treg | 2 | −/+* | Death |
| 5236 | M | 62 | MDS/MPN | BCNU/Melph/Flu | URD | FK/MMF | 2 | −/+† | IR |
| 4986 | M | 55 | MDS | Bu/Cy | URD | FK/MTX | 2 | +/+† | CR |
| 4998 | M | 53 | NHL | BCNU/VP-16/Cy | MRD | FK/PSE | 2 | −/− | CR |
| 5005 | F | 55 | CLL | TLI/ATG | URD-1 | CSA/MMF | 2 | −/+ | No steroids, CR |
| 5182 | F | 60 | Per. T-cell lymphoma | Bu/Cy | URD | FK/MTX | 2 | −/− | CR |
| 5199 | M | 39 | CML blast crisis | TBI/Cy | URD | FK/MTX | 2 | −/+* | CR |
| 5215 | M | 66 | CLL | TLI/ATG | URD | CSA/MMF | 3 | −/− | CR |
| 5261 | M | 53 | ALL | TBI/Cy | URD | FK/MTX | 3 | −/− | CR |
| 5314 | F | 38 | ALL | TBI/VP-16/Cy | URN-1 | FK/MTX | 2 | +/+ | CR |
ALL, acute lymphoblastic leukemia; AML, acute myeloid leukemia; CLL, chronic lymphocytic leukemia; CML, chronic myeloid leukemia; CR, complete response; CSA, cyclosporine; FK, tacrolimus; Haplo, haploidentical donor; MDS, myelodysplastic syndrome; MMF, mycophenylate mofetil; MPN, myeloproliferative neoplasm; MRD, matched related donor; MTX, methotrexate; NHL, non-Hodgkins lymphoma; IR, incomplete response; PSE, prednisone; URD, unrelated donor; URD-1, unrelated donor with 1 mismatch.
Underlined patients are SR; all others are PR.
CMV PCR tissue positivity <150 copies, shell vial, and culture negative.
CMV PCR blood positivity.

Measurement of T-cell repertoire oligoclonality in human clinical GVHD samples. (A) Diagram of sample collection for patients. (B) GI tissue samples from primary-responsive (PR; n = 8) and steroid-refractory (SR; n = 7) patients show the same - cell repertoire oligoclonality as measured by the Gini coefficient (Mann-Whitney, P = .5893). Comparison is of patient averages of individual tissue samples. (C) Blood samples from PR and SR patients at time of diagnosis (●) and day +30 after diagnosis (○) show the same T-cell repertoire oligoclonality (Mann-Whitney, P = .1919). (D) GI tissue samples do not show increased T-cell repertoire oligoclonality with increased GVHD histology grade. Comparison is of average Gini coefficients per patient at each histology grade (Kruskal-Wallis, P = .2600). (E) No significant change in oligoclonality in the blood between the day of diagnosis and day +30 after diagnosis for either PR or SR patients (Wilcoxon, P = .2188).
Measurement of T-cell repertoire oligoclonality in human clinical GVHD samples. (A) Diagram of sample collection for patients. (B) GI tissue samples from primary-responsive (PR; n = 8) and steroid-refractory (SR; n = 7) patients show the same - cell repertoire oligoclonality as measured by the Gini coefficient (Mann-Whitney, P = .5893). Comparison is of patient averages of individual tissue samples. (C) Blood samples from PR and SR patients at time of diagnosis (●) and day +30 after diagnosis (○) show the same T-cell repertoire oligoclonality (Mann-Whitney, P = .1919). (D) GI tissue samples do not show increased T-cell repertoire oligoclonality with increased GVHD histology grade. Comparison is of average Gini coefficients per patient at each histology grade (Kruskal-Wallis, P = .2600). (E) No significant change in oligoclonality in the blood between the day of diagnosis and day +30 after diagnosis for either PR or SR patients (Wilcoxon, P = .2188).
Compared with PR patients, SR patients showed no significant difference in stool output, aGVHD grade (Mann-Whitney, P = .577), maximum GI histology grade (Mann-Whitney, P = .27), or white blood cell count at endoscopy (data not shown; t test, P = .5). All patients received blood-mobilized peripheral stem cells, and the 2 groups showed rough equivalency between numbers of matched related, unrelated, and mismatched donors, with the exception of 1 patient in the SR group who had a haploidentical donor.
Notable differences between groups include 2 patients in the PR group who received nonmyeloablative conditioning but had full donor chimerism at the time of endoscopy. Two patients in the SR group underwent prophylactic treatment with experimental T-regulatory cell therapy. Two patients had CMV PCR positivity at <150 copies with negative shell vial and CMV culture results from the GI biopsy; they were among 4 patients treated for CMV viremia with gancyclovir without evidence of end-organ involvement (Table 1). Some patients had skin aGVHD; most had this prior to the onset of GI symptoms (supplemental Table 1).
TCRβ repertoires in endoscopic tissue samples have similar characteristics in SR and PR patients
We performed repertoire sequencing of TCRβ CDR3 regions from genomic DNA to analyze 2 to 4 GI biopsies from each patient at the time of diagnosis of suspected GI aGVHD. In each sample, between 6739 and 1 141 229 total productive translated amino acid sequences were obtained after a conservative background subtraction, with no statistically significant difference in sequencing depth between groups (supplemental Table 2; Mann-Whitney, P = .2785). Normal healthy controls run in triplicate showed Pearson correlation coefficients between 0.90 and 0.95, with Bhattacharya coefficient (BC) values between 0.7 and 0.88.
Between groups, GI tissue repertoires did not show a statistically significant difference in oligoclonality, as measured using the Gini coefficient19,20 (Figure 1B; Mann-Whitney, P = .6). We also found that within patients, areas of higher-grade histology did not show any difference in oligoclonality of TCRβ repertoires compared with areas of lower-grade histology (Figure 1D; Kruskall-Wallis, P = .3). These data suggest that oligoclonality of tissue-infiltrating TCRβ repertoires is not an indicator of aGVHD outcomes.
aGVHD tissue samples do not share public T-cell clonotypes
We performed comparisons between tissue samples across all patients and found that there were not unique CDR3 sequences present in all tissues. Nor were public clones found in all tissues of either the SR or PR groups. Patients with shared HLA alleles did not appear to show a tendency toward more shared T-cell clones (data not shown). This suggests that tissue-infiltrating TCRβ repertoires are highly patient specific.
T-cell repertoire is more similar across different tissue sites in patients with SR vs PR GVHD
For all same-patient tissue-tissue pairs, we calculated the BC to measure pairwise similarity of TCR repertoires18 (Figure 2A). When averaging tissue-tissue BC values by patient, the intrapatient similarities between colon and upper GI were higher in SR patients than in PR patients (Figure 2B; Mann-Whitney, P = .045). We saw no statistically significant differences in intrapatient colon-colon or upper GI-upper GI similarities (Figure 2B), although these results are for few comparisons.

Similarity of TCR repertoires between tissue samples within patients. (A) Comparison of TCR repertoire similarity by tissue comparison shown for each patient (blue, colon to colon; pink, upper to colon; green, upper to upper). (B) Within patients, the TCR repertoire between upper GI and colon tissue shows greater similarity in SR patients than in PR patients. Results are reported as the average Bhattacharrya coefficient of upper vs colon comparison per patient with upper and lower samples available (for PR, n = 8; for SR, n = 5; Mann-Whitney, P = .045). (C) SR tissue samples are more similar than PR. The best-fit model according to the Akaike information criterion included 2 fixed covariates (patient group and disparity in histology) and fit the data significantly better than a null model containing no fixed covariates (P = .0323).
Similarity of TCR repertoires between tissue samples within patients. (A) Comparison of TCR repertoire similarity by tissue comparison shown for each patient (blue, colon to colon; pink, upper to colon; green, upper to upper). (B) Within patients, the TCR repertoire between upper GI and colon tissue shows greater similarity in SR patients than in PR patients. Results are reported as the average Bhattacharrya coefficient of upper vs colon comparison per patient with upper and lower samples available (for PR, n = 8; for SR, n = 5; Mann-Whitney, P = .045). (C) SR tissue samples are more similar than PR. The best-fit model according to the Akaike information criterion included 2 fixed covariates (patient group and disparity in histology) and fit the data significantly better than a null model containing no fixed covariates (P = .0323).
To comprehensively examine factors that might influence intrapatient tissue-tissue similarity, we fit all intrapatient tissue-tissue BC data to several linear mixed-effects models by maximum likelihood. The models included combinations of the following fixed effects: patient group (SR vs PR), tissue site (upper GI vs colon), and disparity in tissue histology (same histology vs different; 0-5, given 6 possible histology grades: normal, apoptotic bodies, or GVHD grades 1-4).
The best-fit model according to the Akaike information criterion included 2 fixed covariates (patient group and disparity in histology) and fit the data significantly better than a null model containing the random effect but no fixed covariates (P = .032). The combined model (3 fixed covariates) did not fit the data significantly better than the 2 fixed covariates model. This model predicts a BC of 0.34 for a pair of tissues from a PR patient that have the same histology; pairs from a SR patient are predicted to be more similar (BC, +0.21) and those with different histology to be less similar (BC, −0.097). These findings may indicate that patients with severe SR GI aGVHD show a more restricted reconstitution of the entire GI tract, with the possibility that a common suite of aGVHD-causing T-cell clones affect multiple tissue sites within a patient.
Whole TCRβ repertoires in blood do not distinguish SR from PR patients at time of endoscopic evaluation
Because repeated endoscopic biopsies are prohibitive, we cannot directly monitor alloreactive clones in the GI tract, which likely expand as aGVHD worsens. We therefore investigated whether characteristics of TCRβ repertoires in peripheral blood alone distinguish the 2 groups of patients. T-cell repertoire sequencing of blood samples yielded 9193 to 2 196 016 total sequence reads per sample, with no statistically significant difference between groups (supplemental Table 2; Mann-Whitney, P = .37). Oligoclonality in blood at the time of diagnosis as measured by the Gini coefficient was not statistically different between patient groups (Figure 1C; Mann-Whitney, P = .19). Neither group showed significant changes in oligoclonality between time of diagnosis and day 30 post-Dx (Figure 1E; Wilcoxon: PR, P > .99; SR, P = .22). We were also unable to distinguish between the groups by difference in Gini coefficient from time of diagnosis to day 30 post-Dx (data not shown; Mann-Whitney, P = .39).
Relative frequency at diagnosis and longitudinal expansion of aGVHD indicator clones in blood correlates with SR disease
We hypothesized that a blood-derived signature that follows patient-specific, dominant “indicator clones” identified in GI tissue samples could distinguish between SR and PR patients. We define indicator clones as the 100 most frequent clones in a GI tissue sample. We developed a novel “fence plot” to depict indicator clone frequency and rank in the tissue and blood samples of a patient with severe SR aGVHD (Figure 3A) and another patient with PR aGVHD (Figure 3B). The number of tissue indicator clones detected in the blood at the time of diagnosis is shown in Figure 3C. At the time of diagnosis, the number of indicator clones detected in the blood was indistinguishable between groups (Mann-Whitney, P = .22). Similarly, no statistically significant difference in mean or median indicator clone frequency in the blood at the time of diagnosis could be detected between groups (data not shown; Mann-Whitney: mean, P = .33; median, P = .65).

T-cell clonal dynamics of GI-identified TCR indicator clones in the blood. (A) Fence plot illustrating change in frequency and rank of GI indicator clones in a representative patient with SR GI aGVHD (patient 5026). Each clone is represented in rank order (most frequent on top) on a sample’s axis by a node with size proportional to the clone’s frequency in the sample. The sample axis represents 100% of the clone counts above background, so the width of a node corresponds to the percent frequency of the clone in the sample. Clones are colored based on whether they show an increase (orange) or decrease (blue) in frequency in blood at day 30 post-Dx vs at time of diagnosis. Clones of rare frequency that were detected below background or were undetected are depicted off-axis as <bkgd or ND, respectively. (B) Fence plot of a representative patient with PR GI aGVHD (patient 4998). (C-D) Number of indicator clones detected in blood of patients at (C) time of diagnosis and (D) day 30 post-Dx. Points represent indicator clone number for individual tissue biopsies. The same number of GI indicator clones are discovered among PR vs SR patients at the time of diagnosis (C; Mann-Whitney, P = .2174). More GI-GVHD indicator clones are detected in blood at day 30 post-Dx in SR compared with PR patients (Mann-Whitney, P < .0001).
T-cell clonal dynamics of GI-identified TCR indicator clones in the blood. (A) Fence plot illustrating change in frequency and rank of GI indicator clones in a representative patient with SR GI aGVHD (patient 5026). Each clone is represented in rank order (most frequent on top) on a sample’s axis by a node with size proportional to the clone’s frequency in the sample. The sample axis represents 100% of the clone counts above background, so the width of a node corresponds to the percent frequency of the clone in the sample. Clones are colored based on whether they show an increase (orange) or decrease (blue) in frequency in blood at day 30 post-Dx vs at time of diagnosis. Clones of rare frequency that were detected below background or were undetected are depicted off-axis as <bkgd or ND, respectively. (B) Fence plot of a representative patient with PR GI aGVHD (patient 4998). (C-D) Number of indicator clones detected in blood of patients at (C) time of diagnosis and (D) day 30 post-Dx. Points represent indicator clone number for individual tissue biopsies. The same number of GI indicator clones are discovered among PR vs SR patients at the time of diagnosis (C; Mann-Whitney, P = .2174). More GI-GVHD indicator clones are detected in blood at day 30 post-Dx in SR compared with PR patients (Mann-Whitney, P < .0001).
However, at day 30 post-Dx, fewer indicator clones were detected in the blood of PR patients than in SR patients (Figure 3D; Mann-Whitney, P < .0001). We hypothesized that indicator clones in the peripheral blood of SR patients would increase in frequency over time but remain the same or decrease in frequency in PR patients.
We next examined the behavior of GI indicator clones for each patient and plotted the ratio fold change of indicator clone frequencies at day 30 post-Dx vs time of diagnosis (Figure 4A). We observed that a number of indicator clones in SR subjects expanded 10- to 100-fold, but none did in the PR group. To evaluate this further, we devised the ICI-30 as a metric of expansion or contraction of tissue-identified indicator clones in peripheral blood at day 30 post-Dx compared with at diagnosis.

Change in frequency of GI indicator clones between diagnosis and day +30 after diagnosis. (A) SR patients but not PR patients show a longitudinal expansion of GI indicator clones in the blood (top 100 clones in GI tissue). Shown are all GI indicator clones that were detected in blood, reported as ratio fold change of indicator clones at day +30 after diagnosis compared with time of diagnosis. As measured by ICI-30, SR patients show a statistically significant expansion of GI indicator clones when comparing (B) tissue samples or (C) averaged tissues per patient. (D) Control ICI-30 values calculated by defining indicator clones as the 100 most frequent clones in blood at time of diagnosis are not different between PR and SR patients.
Change in frequency of GI indicator clones between diagnosis and day +30 after diagnosis. (A) SR patients but not PR patients show a longitudinal expansion of GI indicator clones in the blood (top 100 clones in GI tissue). Shown are all GI indicator clones that were detected in blood, reported as ratio fold change of indicator clones at day +30 after diagnosis compared with time of diagnosis. As measured by ICI-30, SR patients show a statistically significant expansion of GI indicator clones when comparing (B) tissue samples or (C) averaged tissues per patient. (D) Control ICI-30 values calculated by defining indicator clones as the 100 most frequent clones in blood at time of diagnosis are not different between PR and SR patients.
We found a statistically significant expansion of indicator clones in patients with SR GI aGVHD compared with PR patients, as measured by higher ICI-30 tissue values (Figure 4B; Mann-Whitney, P < .0001). Averaging all tissue ICI-30 values for a patient also showed a statistically significant difference between groups (Figure 4C; Mann-Whitney, P = .007). Although day 30 after diagnosis is late in disease progression and intervention, we performed a receiver operator curve analysis as an early evaluation of eventual clinical feasibility (area under the curve = 0.910, P < .008; supplemental Figure 1). As a control, we calculated ICI-30 values for each patient, defining indicator clones as the top 100 clones by frequency in blood at time of diagnosis (Figure 4D). Control ICI-30 values for SR and PR groups were statistically indistinguishable (Mann-Whitney, P = .094).
We broadened our definition of indicator clone to include any clone above background in the GI tissue biopsy, and there was still a statistically significant difference (supplemental Figure 2; Mann-Whitney, P = .021). Last, we found that steroid dose does not correlate with ICI-30 values for either patient group (data not shown; linear regression: PR, R2 = 0.031, P = .68; SR: R2 = 0.23, P = .28).
Taken together, our data suggest that longitudinal expansion of tissue-infiltrating clones in blood is a biomarker of aGVHD disease activity that can be quantified and used to discriminate outcomes.
Discussion
This study is the first to use comprehensive TCR sequencing to evaluate the T-cell repertoire of patients with aGVHD following allogeneic HCT for treatment of hematological malignancies. GI aGVHD carries a high mortality, and it cannot yet be anticipated or well stratified by symptomology or histology at the time of diagnosis. An additional challenge in the treatment of GI aGVHD is that no objective immune system metrics exist that can be used to guide therapy. We hypothesized that TCRβ repertoire sequencing might identify patterns that could eventually be used to predict severity and/or track the progress of GI aGVHD.
Because T cells are known to drive the pathophysiology of GVHD, assessment of T-cell repertoires is likely to directly reflect disease activity. Sophisticated immune monitoring of this type may allow greater clarity into the pathophysiology of aGVHD, as well as provide much-needed objective measures for guiding therapy and standardizing patients in clinical studies. Further, it may eventually be possible to deduce the identity of the target antigens through analysis of the TCR repertoire. We used repertoire sequencing to identify dominant personal T-cell clones in the GI tracts of patients with GI aGVHD symptoms at the time of diagnosis and to track their behavior in blood over time. Importantly, we demonstrated that this technology is applicable to the study of a complex disease such as GVHD, and analysis of personalized TCR repertoire is readily achievable.
Repertoire sequencing is subject to the introduction of errors and artifacts due to primer dimerization, contamination, polymerase errors, and PCR amplification biases. We controlled for some potential technical errors by using conservative background subtraction to minimize the contribution of artifacts. We also designed our analysis to minimize any effect of PCR amplification bias, specifically by following the relative change in frequency of indicator clones in samples from the same patient.
Our study is the first to demonstrate a number of key points. First, despite the fact that PR and SR patients showed the same degree of oligoclonality in their tissue samples at the time of diagnosis (Figure 1B), we found that patients with SR GVHD appeared to have a more consistent TCRβ clonal structure between different biopsy sites in the GI tract than PR counterparts (Figure 2). The high similarity of T-cell repertoires between upper and lower GI samples could be used as a way to predict and stratify SR vs steroid-responsive disease at the time of diagnosis. This finding could also suggest that SR patients may have a unique pathology. For example, antigens targeted by common T-cell clones within patients with severe, SR disease might not necessarily be unique to specific areas of the GI tract, as may be the case for PR patients. These potential common allogeneic antigens may be present and/or targeted throughout the GI tract and at other common aGVHD sites, such as the skin and liver. Future studies will require standardized collection of skin or liver sites in addition to multiple sites throughout the GI tract.
A second major finding in our study is that longitudinal tracking of these GI-identified clones in peripheral blood revealed an increased clonal expansion in patients with SR disease vs PR patients. When we tracked GI-identified aGVHD indicator clones in blood from the time of diagnosis, we were able to detect a sizable number of these clones, but at 30 days after diagnosis, we detected fewer indicator clones in the blood of PR patients. We expect that in successfully treated cases, aGVHD-associated clones that presumably decrease in frequency would be less likely to be detected in the blood.
There are a number of potential confounding factors that distinguish the SR group, including the fact that most patients in the SR group received etanercept, and 2 patients in the group received experimental T-regulatory cells. Neither of these factors is associated with inducing clonal expansion to the best of our knowledge. Alternatively, patients with SR GVHD and on more intense immunosuppression could have the proliferation of gut-associated T cells due to infectious causes such as gut translocation of bacteria. The 1 SR patient with an ICI-30 score similar to those of the 8 PR patients had CMV PCR positivity. The potential confounding effects of CMV viremia warrants further investigation.
We do not know how many T-cell clones mediate human GI aGVHD. In some patients, a few clones could cause the disease, whereas in others, hundreds might be responsible. Based on our tracking of aGVHD indicator clones, a relatively small number of GI-identified clones expanded dramatically (Figure 4A); our data support the use of repertoire sequencing to define and quantify aGVHD-inducing clones in a personalized fashion.
A third major finding of our study was that no clones were shared between all patients with aGVHD, indicating that there are no universal TCRβ sequences that cause aGVHD. Although 1 interpretation of this finding is that there exists no universal aGVHD allogeneic antigen, it is also possible that multiple TCRβ CDR3 sequences may recognize the same antigen. Our study is on a small group, and evaluating more patients who share HLA alleles could still yield dominant shared clonotypes and other reproducible patterns.
A fourth major point is that it was absolutely essential to biopsy and study clones identified in GI tissue affected by GVHD to accurately classify patients. Over time, it might be possible to use chemokine or trafficking receptor selection of T cells to profile clones targeting specific tissue sites, but until then, studying peripheral blood in isolation from tissue is not likely to be productive.
Further investigation will be needed to determine whether expansion of GI-identified clones can be detected 7 to 14 days after diagnosis, a critical time point at which stratification of SR patients is clinically relevant. The single patient who survived SR GI aGVHD had an ICI-30 value at the very lowest end of the SR group. This might suggest that our approach could someday be used to titrate therapy. Whether this approach is also applicable to autoimmune disorders warrants investigation.
In summary, our study is the first to use repertoire sequencing to assess the dynamics and complexity of the T-cell repertoire in GI aGVHD over time and offers a new approach to the study of immune function and disease in humans.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported by the National Institutes of Health, National Cancer Institute P01 CA049605 NIH/National Cancer Institute. E.H.M. was supported by the American Society for Blood and Marrow Transplantation Young Investigator award and a Stanford Translational and Applied Medicine Pilot grant (Stanford Department of Medicine).
Authorship
Contribution: E.H.M. designed the experiments and clinical protocol, contributed to analysis, and authored the paper; A.R.H., A.L., D.B.M., D.S.J., and P.L. contributed to analysis and writing; E.H.M., J.L., M.F., and J.L.Z. contributed to sample collection and preparation; and R.S.N. and S.S. contributed to design of protocol and writing.
Conflict-of-interest disclosure: E.H.M. and A.R.H. have equity in GigaGen, Inc. A.L. and D.S.J. have equity with and are employed by GigaGen, Inc. The remaining authors declare no competing financial interests.
Correspondence: Robert Negrin, Division of Blood and Marrow Transplantation, Stanford Medical School, Stanford University, CCSR, Room 2205, 269 W. Campus Dr, Stanford, CA 94305; e-mail: negrs@stanford.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal