

Abstract
Noninvasive monitoring of minimal residual disease (MRD) has led to significant advances in personalized management of patients with hematologic malignancies. Improved therapeutic options and prolonged survival have further increased the need for sensitive tumor assessment that can inform treatment decisions and patient outcomes. At diagnosis or relapse of most hematologic neoplasms, malignant cells are often easily accessible in the blood as circulating tumor cells (CTCs), making them ideal targets to noninvasively profile the molecular features of each patient. In other cancer types, CTCs are generally rare and noninvasive molecular detection relies on circulating tumor DNA (ctDNA) shed from tumor deposits into circulation. The ability to precisely detect and quantify CTCs and ctDNA could minimize invasive procedures and improve prediction of clinical outcomes. Technical advances in MRD detection methods in recent years have led to reduced costs and increased sensitivity, specificity, and applicability. Among currently available tests, high-throughput sequencing (HTS)–based approaches are increasingly attractive for noninvasive molecular testing. HTS-based methods can simultaneously identify multiple genetic markers with high sensitivity and specificity without individual optimization. In this review, we present an overview of techniques used for noninvasive molecular disease detection in selected myeloid and lymphoid neoplasms, with a focus on the current and future role of HTS-based assays.
Introduction
For any given malignancy, the success of disease detection from peripheral blood (PB; ie, noninvasive disease detection) generally requires several considerations: knowledge of tumor biology and its molecular characteristics, accessibility of tumor material in circulation, and technical factors. Most hematologic neoplasms are circulating cancers; therefore, circulating tumor cells (CTCs) are often easily accessible in the blood at times of overt disease. Other malignancies, including lymphomas, often manifest in noncirculating tissue compartments and organs, thus making CTCs rare. Here, circulating tumor DNA (ctDNA) in the bloodstream can be used for noninvasive tumor detection (Tables 1 and 2; Figure 1).1,2 Unfortunately, the fraction of CTCs and ctDNA in the pool of normal PB mononuclear cells (PBMCs) or cell-free DNA is often very small. Thus, the analytical sensitivity and specificity of a noninvasive test must be sufficiently high to detect minimal tumor loads that almost invariably lead to future disease progression. Furthermore, to minimize costs and allow clinical implementation, a test should apply to a wide range of patients without patient-specific optimization.
Clinical impact of noninvasive disease detection at distinct disease milestones in lymphoid malignancies
| Diagnosis | Precancerous condition | Diagnosis/pretreatment | During therapy | Posttreatment/surveillance | Relapse/progression |
|---|---|---|---|---|---|
| ALL | CTC or malignant BM cell: levels predict outcome before allo-SCT38,41,43,55 | CTC or malignant BM cell: positivity predicts clinical outcome29-35,38-43,54,55 ; positivity predicts relapse39,56 ; positivity might guide treatment decisions33,35 | Malignant BM cell: positivity identifies relapse39 | ||
| HL | ctDNA: positivity tends to predict clinical outcome66,68 | ||||
| MM | CTC or malignant BM cell: genotyping defines clinical risk (eg, t(4:14))71 Autograft: positivity predicts clinical outcome82 | Malignant BM cell: positivity predicts clinical outcome72,73,77-79 ; positivity pre-lenalidomide maintenance predicts clinical outcome78 | |||
| MCL | CTC or malignant BM cell: positivity before auto-SCT predicts survival87 | CTC or malignant BM cell: positivity predicts clinical outcome85,86,88 ; detection might guide treatment decisions84 | |||
| CLL | CTC: genotyping defines clinical risk (eg, del(17p13), TP53)92,93 ; assessment might identify therapeutic targets (eg, del(17p13))92,93 | CTC or malignant BM cell: levels predict clinical outcome68,94-99,103,104 ; dynamics predict relapse97 | |||
| FL | Healthy PBMC: BCL2-IGH rearrangement levels >0.01% are associated with a 23-fold higher risk for malignant transformation112 | ctDNA or malignant BM cells: levels correlate with clinical outcome122,132 | CTC or malignant BM cells: positivity predicts clinical outcome116-120,125,127,129,131 | CTC: positivity identifies relapse116 | |
| DLBCL | ctDNA: levels correlate with tumor burden and PFS22,134,135 ; genotyping identifies COO subtypes22 ; profiling identifies DH and TH lymphomas22 | ctDNA: profiling identifies resistant clones22,137 ; negativity after 2 cycles predicts PFS134 | CTC or ctDNA: predicts relapse with 3-6 month lead time22,134,135 ; positivity predicts clinical outcome22,134 | CTC or ctDNA: positivity identifies relapse22,134,135,137 ; profiling detects histological transformation22 |
| Diagnosis | Precancerous condition | Diagnosis/pretreatment | During therapy | Posttreatment/surveillance | Relapse/progression |
|---|---|---|---|---|---|
| ALL | CTC or malignant BM cell: levels predict outcome before allo-SCT38,41,43,55 | CTC or malignant BM cell: positivity predicts clinical outcome29-35,38-43,54,55 ; positivity predicts relapse39,56 ; positivity might guide treatment decisions33,35 | Malignant BM cell: positivity identifies relapse39 | ||
| HL | ctDNA: positivity tends to predict clinical outcome66,68 | ||||
| MM | CTC or malignant BM cell: genotyping defines clinical risk (eg, t(4:14))71 Autograft: positivity predicts clinical outcome82 | Malignant BM cell: positivity predicts clinical outcome72,73,77-79 ; positivity pre-lenalidomide maintenance predicts clinical outcome78 | |||
| MCL | CTC or malignant BM cell: positivity before auto-SCT predicts survival87 | CTC or malignant BM cell: positivity predicts clinical outcome85,86,88 ; detection might guide treatment decisions84 | |||
| CLL | CTC: genotyping defines clinical risk (eg, del(17p13), TP53)92,93 ; assessment might identify therapeutic targets (eg, del(17p13))92,93 | CTC or malignant BM cell: levels predict clinical outcome68,94-99,103,104 ; dynamics predict relapse97 | |||
| FL | Healthy PBMC: BCL2-IGH rearrangement levels >0.01% are associated with a 23-fold higher risk for malignant transformation112 | ctDNA or malignant BM cells: levels correlate with clinical outcome122,132 | CTC or malignant BM cells: positivity predicts clinical outcome116-120,125,127,129,131 | CTC: positivity identifies relapse116 | |
| DLBCL | ctDNA: levels correlate with tumor burden and PFS22,134,135 ; genotyping identifies COO subtypes22 ; profiling identifies DH and TH lymphomas22 | ctDNA: profiling identifies resistant clones22,137 ; negativity after 2 cycles predicts PFS134 | CTC or ctDNA: predicts relapse with 3-6 month lead time22,134,135 ; positivity predicts clinical outcome22,134 | CTC or ctDNA: positivity identifies relapse22,134,135,137 ; profiling detects histological transformation22 |
Role of PBMC, CTC, ctDNA, and BM cell profiling for detection of premalignant states in healthy individuals, identification of clinically relevant biomarkers, and prediction of outcome in lymphoid malignancies at distinct disease milestones.
COO, cell of origin; DH, double hit; DLBCL, diffuse large B-cell lymphoma; TH, triple hit.
Clinical impact of noninvasive disease detection at distinct disease milestones in myeloid malignancies
| Diagnosis | Precancerous condition | Diagnosis/pretreatment | During therapy | Posttreatment/surveillance | Relapse/progression |
|---|---|---|---|---|---|
| AML | Healthy PBMC: harbor age-dependent aberrations associated with overt AML/MDS138-142 | CTC or malignant BM cell: genotyping defines molecular prognostic factors150,151 ; genotyping might identify therapeutic targets (eg, FLT3)151,152 | CTC or malignant BM cell: positivity and kinetics during therapy predict risk of relapse155,157 | CTC or malignant BM cell: positivity postinduction and postconsolidation predicts clinical outcome156,157,161,164 | CTC or malignant BM cell: profiling identifies emergent clones at relapse163 ; profiling identifies relapse161 |
| CML | CTC or malignant BM cell: BCR-ABL1 levels predict clinical outcome168-170 ; profiling identifies resistance mutations176,177,180,181 | CTC or malignant BM cell: rising BCR-ABL1 levels indicate progression178 | |||
| MPN | CTC or malignant BM cell: genotyping is part of diagnostic criteria189 | CTC or malignant BM cell: levels predict clinical outcome190,191 |
| Diagnosis | Precancerous condition | Diagnosis/pretreatment | During therapy | Posttreatment/surveillance | Relapse/progression |
|---|---|---|---|---|---|
| AML | Healthy PBMC: harbor age-dependent aberrations associated with overt AML/MDS138-142 | CTC or malignant BM cell: genotyping defines molecular prognostic factors150,151 ; genotyping might identify therapeutic targets (eg, FLT3)151,152 | CTC or malignant BM cell: positivity and kinetics during therapy predict risk of relapse155,157 | CTC or malignant BM cell: positivity postinduction and postconsolidation predicts clinical outcome156,157,161,164 | CTC or malignant BM cell: profiling identifies emergent clones at relapse163 ; profiling identifies relapse161 |
| CML | CTC or malignant BM cell: BCR-ABL1 levels predict clinical outcome168-170 ; profiling identifies resistance mutations176,177,180,181 | CTC or malignant BM cell: rising BCR-ABL1 levels indicate progression178 | |||
| MPN | CTC or malignant BM cell: genotyping is part of diagnostic criteria189 | CTC or malignant BM cell: levels predict clinical outcome190,191 |
Role of PBMC, CTC, ctDNA, and BM cell profiling for detection of premalignant states in healthy individuals, identification of clinically relevant biomarkers, and prediction of outcome in myeloid malignancies at distinct disease milestones.

DNA sources for MRD detection in hematologic malignancies. Graphic overview of distinct DNA sources for molecular disease profiling in hematologic neoplasms, which include circulating tumor DNA as part of the circulating cell-free DNA pool and circulating tumor cells in the bloodstream as well as malignant cells in the BM compartment. Their main characteristics and role in hematologic cancers are displayed in the table next to the graphic.
DNA sources for MRD detection in hematologic malignancies. Graphic overview of distinct DNA sources for molecular disease profiling in hematologic neoplasms, which include circulating tumor DNA as part of the circulating cell-free DNA pool and circulating tumor cells in the bloodstream as well as malignant cells in the BM compartment. Their main characteristics and role in hematologic cancers are displayed in the table next to the graphic.
Another important consideration is the type of genetic aberration being detected. Some malignancies are characterized by a single stereotypic genetic variant, which can be assessed in nearly every patient by highly sensitive approaches.3,4 Molecular aberrations in other hematological neoplasms are heterogeneous, and detailed knowledge of the underlying genetic landscape is required. Finally, sensitivity strongly relies on input material quantity and quality. For example, a single gene test can only achieve a sensitivity of 1 in 20 000 if the input material matches or exceeds this threshold (Figure 2).5

Detection limits of methods used for DNA identification in PB and BM. Diagrams depicting the range of detection of methods used for identification of cellular DNA in PB and BM (A) and cell-free DNA in plasma (B). PB input material for each assay was considered from a normal 10 mL EDTA vacutainer. aAnalytical sensitivity highly depends on panel width, sequencing depth, and technical conditions (eg, barcoding, duplex sequencing). bAnalytical sensitivity substantially varies depending on the target/targets interrogated and number of input genomes. cAnalytical sensitivity depends on the method used and number of markers tested. WES, whole exome sequencing; WGS, whole genome sequencing.
Detection limits of methods used for DNA identification in PB and BM. Diagrams depicting the range of detection of methods used for identification of cellular DNA in PB and BM (A) and cell-free DNA in plasma (B). PB input material for each assay was considered from a normal 10 mL EDTA vacutainer. aAnalytical sensitivity highly depends on panel width, sequencing depth, and technical conditions (eg, barcoding, duplex sequencing). bAnalytical sensitivity substantially varies depending on the target/targets interrogated and number of input genomes. cAnalytical sensitivity depends on the method used and number of markers tested. WES, whole exome sequencing; WGS, whole genome sequencing.
In hematologic neoplasms, several technologies have been established for noninvasive minimal residual disease (MRD) detection and response assessment: flow cytometry (eg, multicolor flow cytometry [MFC]), polymerase chain reaction (PCR)-based methods that do not rely on sequencing (quantitative real-time PCR [qPCR], digital PCR [dPCR], reverse transcription PCR [RT-PCR]), and high-throughput sequencing (HTS, next-generation sequencing [NGS]) methods, which allow massive parallel sequencing of DNA molecules in a single flow cell and produce millions or billions of sequences concurrently (Figure 2; Table 3). For balancing brevity with clarity in this review, we focus on increasingly common HTS approaches and encourage readers to consult dedicated sources for MRD methods that do not rely on HTS.6-11
Key characteristics of methods used for noninvasive disease detection in hematologic malignancies
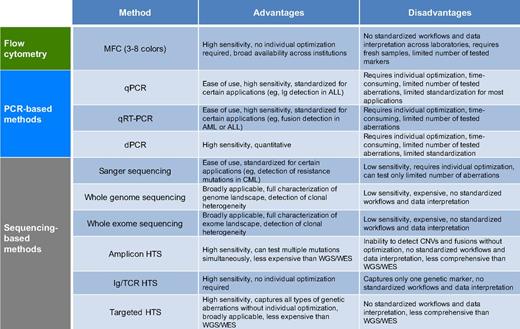 |
|
Colors in column 1 refer to the colors in Figure 2.
CNVs, copy number variations; WES, whole exome sequencing; WGS, whole genome sequencing.
HTS methods are powerful tools for noninvasive disease detection with several dedicated advantages over other approaches. They can detect the entire spectrum of genetic alterations, including single nucleotide variants, insertions/deletions, chromosomal rearrangements, and copy number changes, and thus can overcome the limitations of assays covering single somatic variants (Table 3).12-14 Moreover, HTS assays are usually applicable to a broader population of patients and patient-specific optimization or knowledge of tumor genotypes is generally not required. To achieve sequencing depths that allow sensitive noninvasive disease detection similar to modern qPCR or dPCR assays (0.001% to 0.0001% allele frequency [AF]), targeted approaches are generally preferred over whole genome or exome sequencing, especially when considering current costs and sequencing error rates. These targeting methods include amplicon-based or targeted hybrid-capture techniques to select and enrich the genomic regions of interest.15-18 The ability to simultaneously detect and monitor multiple aberrations and recent methodological advances in reducing errors have improved analytical sensitivity of targeted HTS assays down to ∼0.0001% (Figure 2).16,19,20 Furthermore, HTS-based assays facilitate assessment of intra- and intertumor heterogeneity, allowing individualized detection of clonal evolution.21,22 Although HTS methods are becoming more common, they are not widely used in clinical laboratories yet and therefore are not prominently featured in disease management guidelines. Methodological challenges, both in molecular biology and bioinformatics analyses, must be overcome as these methods become routinely used.
Noninvasive disease detection in lymphoid malignancies
Acute lymphoblastic leukemia
Lymphoid malignancies are characterized by clonal immunoglobulin (Ig) or T-cell receptor (TCR) gene rearrangements, which take place during early B- and T-cell development. This genetic feature is highly specific to each individual tumor and thus represents an ideal molecular marker for sensitive disease detection by PCR- and HTS-based techniques. Consequently, MRD detection using Ig or TCR rearrangements has become widely adopted in lymphoid cancers.
Acute lymphoblastic leukemia (ALL) is exemplary for how MRD assessment can be incorporated into treatment protocols and clinical guidelines. Nearly 80% to 95% of adult ALL patients and more than 95% of pediatric patients achieve complete remissions (CRs) after induction chemotherapy.23,24 However, a substantial proportion of patients will relapse and ultimately succumb to their disease. Clinical evidence for the utility of MRD in ALL has led to its broad adoption for patient risk stratification and early therapeutic response monitoring. Current standard approaches include flow cytometry (sensitivity, ∼0.01%), allele-specific oligonucleotide (ASO) qPCR of Ig/TCR gene rearrangements (∼0.001%), and qRT-PCR of fusion transcripts (eg, BCR-ABL1, MLL-AF4; 0.01% to 0.001%).8,25-28 Although flow cytometry and ASO-qPCR are applicable to virtually all patients, the use of qRT-PCR is limited to patients with detectable fusions.25 However, MRD status assessed by those techniques is one of the most powerful predictors of survival and guides treatment decisions (Table 1).29-43 For detailed reviews, see van Dongen et al,25 Brüggemann et al,44 and Campana et al.45
All of these methods, despite demonstrating utility in clinical settings, have limitations. For example, they typically rely on invasive bone marrow (BM) sampling and cannot capture clonal heterogeneity.46,47 Modern approaches combining universal PCR primers for rearranged Ig/TCR regions with HTS have the potential to overcome those shortcomings. Proof-of-principle analyses applying this technique to PB and BM samples demonstrated high sensitivity (0.001% to 0.0001%) and high concordance with ASO-qPCR and flow cytometry.48-53 Moreover, it allows identification of oligoclonality at diagnosis and noninvasive disease monitoring over time.49,50 Logan et al,54 Pulsipher et al,55 and Mannis et al56 demonstrated that MRD assessment by this HTS approach accurately predicts relapse and patient survival before and after allogeneic stem cell transplantation (allo-SCT) or autologous SCT (auto-SCT), using either noninvasively acquired CTCs or invasive BM biopsies. Additionally, it can provide information on the physiological repertoire of B and T cells, which appears to be prognostically relevant.57,58
However, HTS analysis of BM still appears superior to PB in ALL, possibly from a higher concentration of tumor cells.51,54 Nevertheless, the reported sensitivities of MRD monitoring using less invasive blood samples seems to be promising and is moving forward into future clinical trials.48-51,54 International standardization and validation of noninvasive HTS-based methods in multicenter studies is needed to demonstrate robust clinical utility.
Hodgkin lymphoma
Despite high initial response rates to combination chemotherapy, 20% to 30% of patients with Hodgkin lymphoma (HL) will experience either primary refractoriness to chemotherapy or disease relapse.59 Identifying genetic features for disease monitoring and response assessment has been challenging in this disease, as malignant cells are usually very rare in bulk tissue (<5%). However, by enriching tumor cells through microdissection, some recurrent aberrations could be identified that can be followed for noninvasive MRD monitoring, including mutations in specific genes (eg, TNFAIP3, XPO1) and pathways (NF-κB, JAK/STAT), as well as chromosomal aberrations, translocations, and rearrangements involving Ig.60-66
Camus et al demonstrated that patients with XPO1 mutations detected in plasma by dPCR (0.1%) at the end of standard chemotherapy had a shorter progression-free survival (PFS) than XPO1-negative cases.67 In a large study conducted by Herrera et al, ctDNA positivity assessed by HTS of Ig rearrangements (IgHTS) after allo-SCT predicted clinical outcomes.68 However, Oki et al showed that IgHTS fails to identify Ig clonotypes at diagnosis in more than one-quarter of patients, limiting its use as a ubiquitous biomarker.69 Vandenberghe et al identified chromosomal aberrations by pretreatment ctDNA profiling using whole genome sequencing and observed that normalization of ctDNA after initiation of therapy mirrors treatment response.66
These studies demonstrate feasibility of noninvasive MRD monitoring in HL. However, improvements are needed to increase sensitivity and broaden the applicability of PCR- and HTS-based methods.
Multiple myeloma
Major therapeutic advances in the past decade have significantly improved outcomes of patients with multiple myeloma (MM).70 Consequently, both the role of diagnostic genotyping to define prognostic markers (eg, del(17p13), t(4;14)) and MRD assessment to monitor treatment efficacy has become increasingly important.71 Current standard approaches for MRD detection in clinical trials and research studies include MFC and ASO-qPCR of rearranged Ig regions (0.01% to 0.001%). Both have demonstrated clinical utility: MRD negativity by MFC at day 100 after auto-SCT was associated with improved survival.10,72 Similarly, Ferrero et al showed that patients achieving major MRD response (<0.01%) by PCR-based methods had a favorable prognosis and excellent disease control.73 However, caveats still remain: despite high sensitivity and reasonable turnaround time, both methods are more applicable to invasive BM specimens than PB samples (for detailed reviews, see Mailankody et al74 and Paiva et al75 ).
Approaches using IgHTS in MM can assess MRD in BM aspirates, PBMCs, and cell-free DNA with sensitivities down to 0.0001%.48,49,76 Several studies applying IgHTS to invasive BM biopsies showed improved sensitivity over conventional methods, demonstrated prognostic relevance of MRD detection, and identified substantial oligoclonality at diagnosis with emergence of subclones over time.77-80 On the other hand, studies investigating noninvasive disease detection in PB are relatively rare. In a proof-of-principle analysis, Gimondi et al were able to identify IGH clonotypes in the plasma of 6 MM patients at diagnosis, demonstrating 100% concordance with tumor V(D)J sequences.81 Wee et al analyzed autografts by 3 different methods (IgHTS, qPCR, and dPCR) in patients undergoing auto-SCT. Here, IgHTS demonstrated the highest sensitivity and prognostic value, suggesting a potential future role for noninvasive MRD detection.82
Mantle cell lymphoma
Patients with mantle cell lymphoma (MCL) are genetically characterized by clonal Ig rearrangements and translocation t(11;14) (CCND1/IGH), resulting in overexpression of CCND1 that enables malignant transformation by cell-cycle deregulation.83 Consequently, the gold standard for MRD detection in current clinical trials is qPCR of CCND1/IGH and/or V(D)J rearrangements with sensitivities down to 0.001%. Numerous studies have demonstrated that MRD assessment by qPCR applied to PB or BM samples in MCL after induction therapy is a strong prognostic factor, even guiding therapeutic decisions in response to molecular relapse after transplant.84-87 However, these assays are applicable to only 40% and 90% of all patients.88,89
Similar to ALL and MM, IgHTS has improved noninvasive disease detection compared with qPCR, offering an alternative for MRD detection.48,52 However, the clinical utility of this greater sensitivity needs to be evaluated prospectively before IgHTS potentially replaces standardized qPCR assays as the gold standard in clinical trials. For further reviews covering this topic, see Hoster et al90 and Pott et al.91
Chronic lymphocytic leukemia
Chronic lymphocytic leukemia (CLL) is an incurable disease with a heterogeneous but often indolent course.92 An increased understanding of the genetic landscape has led to significant advances in assessing clinical outcomes and defining therapeutic procedures (eg, del(17p13), TP53).92,93 Furthermore, MRD detection in PB or BM above a threshold of 0.01% by standardized MFC or ASO-qPCR of Ig rearrangements seems to be an independent predictor of survival.94-96 For example, high-risk CLL patients undergoing allo-SCT have a favorable prognosis if they achieved MRD negativity 12 months after transplant.97-100 However, the use of MRD to tailor treatment in CLL in clinical routine is currently discouraged (for detailed reviews, see Wierda et al,100 Thompson et al,101 and Böttcher et al102 ).
Thus far, there have been only a few studies applying IgHTS to PB in CLL. Rawstron et al103 and Logan et al104 reported concordance between IgHTS and MFC or ASO-qPCR, with increased sensitivity. They also demonstrated that MRD detection >0.0001% at 9, 12, and 24 months after allo-SCT in high-risk patients was associated with subsequent relapse. Moreover, doubling of MRD levels within 12 months after transplant was prognostic for relapse.103 This suggests 2 conclusions: first, an increased sampling frequency might help to predict disease progression; and second, the increased sensitivity of IgHTS over conventional methods might be clinically relevant. Larger studies are needed to validate both of these hypotheses.
Yeh et al applied a targeted HTS approach to serial plasma samples of 32 CLL patients, capturing 7 recurrently mutated genes.105 They revealed that noninvasive ctDNA analysis allows detection of different disease compartments and discovery of emerging genomic changes over time (eg, toward Richter's transformation), highlighting the advantages and potential of methods covering multiple genes over single gene assays (Table 3).
Follicular lymphoma
Chromosomal translocations involving BCL2 and IGH (t(14;18)) are present in >90% of follicular lymphoma (FL) patients. This genetic hallmark occurs during B-cell development and gives rise to constitutive expression of the antiapoptotic protein BCL2.106,107 BCL2/IGH translocation is considered the founding event of FL lymphomagenesis, but is not sufficient for malignant transformation.108 Consequently, t(14;18) can be detected at low frequencies in healthy subjects who will never develop FL.109-111 Roulland et al screened prediagnostic blood from a large cohort of healthy individuals for the presence of t(14;18). By applying qPCR, they found that people with t(14;18) >0.01% AF had a 23-fold higher risk for developing FL, suggesting a potential role as a predictive biomarker.112 However, specificity at this threshold was relatively low (96.5%), which limits the clinical utility of BCL2/IGH detection in healthy individuals; therefore, the use of this test outside research studies is not recommended. Additional recurrent driver mutations have recently been described and are thought to represent early initiating events required for malignant transformation.113-115 Yet, these events have not been identified before overt malignancy; therefore, their role in detecting premalignant states is unclear.
Clinically, FL is the most common indolent NHL; however, recurrent relapses and histological transformation into aggressive disease can lead to decreased survival. Conflicting results have been reported as to whether PCR-based methods for detection of BCL2/IGH or Ig rearrangements in BM or PB can be used for prediction of treatment response and outcome at diagnosis or after therapy.12,116-129 The reported methods have several shortcomings and probably contribute to these controversies: most PCR assays for BCL2/IGH identification are restricted to the major breakpoint region and minor cluster region of BCL2; therefore, only a subset of patients (50% to 65%) can be assessed. Detection of Ig rearrangements can also be ineffective in FL because of high rates of somatic hypermutation (SHM), a characteristic of germinal center lymphomas. Alcaide et al developed a digital droplet PCR method targeting recurrently mutated genes in FL and DLBCL and demonstrated in a proof-of-principle study high sensitivity and specificity for noninvasive ctDNA detection.130
Modern HTS-based approaches also use clonal V(D)J rearrangements for noninvasive detection of FL. Pott et al demonstrated good concordance between t(14;18) qPCR and IgHTS for MRD monitoring in PB samples; however, applicability of IgHTS was not optimal because clonotype sequences could not be identified in 25% of patients.131 Sarkozy et al applied IgHTS to tumor biopsies and plasma samples at diagnosis. They identified tumor clonotypes in 83% of diagnostic ctDNA and showed that patients with high levels of ctDNA have a shorter PFS than patients with low levels.132
Both studies again illustrate that ongoing SHM significantly limits the use of V(D)J sequencing in this disease. Better strategies are therefore needed to overcome these problems in FL.
DLBCL
DLBCL is the most common NHL subtype. A hallmark of this disease is its molecular and clinical heterogeneity. Assessment of ctDNA in the plasma or serum of DLBCL patients by HTS- and PCR-based approaches to evaluate therapeutic response and clinical outcome has been extensively studied in recent years. In a proof-of-concept study, Camus et al used both dPCR and amplicon HTS to detect ctDNA in diagnostic and follow-up plasma samples. They showed that both methods were highly concordant and identified ctDNA in 93% of diagnostic plasma samples, with a sensitivity of 0.05%.133
Roschewski et al and our group set out to evaluate the value of ctDNA as a prognostic biomarker in larger cohorts of patients by applying IgHTS to a series of tumor biopsies, diagnostic and follow-up plasma/serum samples as well as CTCs. Both studies showed a detection rate of ∼85% in tumor biopsies and 82% to 92% in pretreatment ctDNA.134,135 Levels of ctDNA correlated with other measures of tumor burden, including lactate dehydrogenase, metabolic tumor volume, and International Prognostic Index. Furthermore, they suggested a prognostic role for monitoring ctDNA during and after therapy. In patients achieving an initial CR, ctDNA was detectable 3 to 3.5 months before clinical relapse, outperforming positron emission tomography/computed tomography.134,135 In addition, patients with undetectable ctDNA on the first day of cycle 3 of therapy had a superior PFS (but not overall survival) compared with patients with positive ctDNA.134 Importantly, disease detection from plasma significantly outperformed detection from CTCs.135 Despite its value as a prognostic tool, IgHTS has several shortcomings. This includes limited sensitivity in low tumor burden settings and reduced applicability because of SHM, leading to difficulty identifying clonotypic sequences. Furthermore, IgHTS monitors only a single genetic marker and cannot capture complex genetic landscapes.
To overcome some of these challenges, our group recently applied Cancer Personalized Profiling by Deep Sequencing, a targeted capture HTS method, to DLBCL. In this effort, we targeted 268 genes to assess the whole variety of different genetic aberrations simultaneously (point mutations, translocations, insertions/deletions, Ig rearrangements) and to improve sensitivity and applicability to ctDNA.22 In a cohort of 92 patients, we identified somatic alterations in 100% of pretreatment ctDNA samples with a specificity of 99.8% when tumor mutation profiles were known (87% when tumor genotypes were unknown). Pretreatment ctDNA levels correlated with clinical measures of tumor burden and were prognostic of patient outcomes. Relapses could be noninvasively detected in 100% of cases with a mean lead time exceeding 6 months, including detection of disease burden as low as 0.003% AF.22,136 Importantly, clinically relevant tumor heterogeneity was observed in plasma. This included emergence of subclones harboring resistance mutations to targeted therapies, identification of mutation patterns that allowed noninvasive classification of DLBCL cell-of-origin subtypes, and discovery of clonal evolution, distinguishing transformation of FL from indolent FL progression.22 Using a related approach focused on genotyping but not optimized for monitoring, Rossi et al targeted 59 recurrently mutated genes for ctDNA analysis.137 In their study, 85% to 95% of patients had detectable ctDNA at diagnosis when tumor genotypes were unknown, with 83% of all tumor mutations identified in pretreatment plasma. Their longitudinal ctDNA analyses also showed emerging mutations at relapse, suggesting evolution of resistant subclones.
These studies demonstrate the great potential of ctDNA assessment at various disease milestones by HTS. We anticipate a growing role of such technologies to complement positron emission tomography/computed tomography imaging and tumor biopsies for precision patient management within the next few years, initially in trials and ultimately as part of clinical routine.
Noninvasive disease detection in myeloid malignancies
Acute myeloid leukemia and myelodysplastic syndrome
Over the past 5 years, systematic genetic profiling of PB cells in healthy individuals by HTS-based methods has led to a growing understanding of potential precancerous conditions in hematologic cancers.138-142 Several research groups have identified an increase of somatic mutations in the blood of individuals without overt signs of a hematologic malignancy and normal blood counts, including DNMT3A, TET2, and ASXL1 mutations.138-142 These variants seem to occur in hematopoietic stem and progenitor cells and confer survival advantage to clones as they expand. Consequently, they can be detected in mature circulating cells with increasing prevalence in older healthy people (up to 20% of people ≥90 years) in a condition called clonal hematopoiesis of indeterminate potential (CHIP; for detailed reviews, see Link et al and Steensma et al).139,143-145
Recurrent mutations related to age-dependent CHIP have also been described in patients with myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML).146,147 This suggests that they represent potential early genetic events in the clonal selection process leading to hematologic cancers. Indeed, by studying whole exome sequencing DNA from nearly 30 000 PB samples, Genovese et al138 and Jaiswal et al139 demonstrated that elderly individuals harboring CHIP-associated mutations have a significantly higher risk for developing hematologic neoplasms (hazard ratio, 11.1; 95% confidence interval, 3.9-32.6) and death (hazard ratio, 1.4; 95% confidence interval, 1.1-1.8). These studies suggest a potential future role of population-based screening assays to identify healthy individuals at risk for malignant transformation.
At the same time, there is evidence that only a minority of persons carrying CHIP-associated mutations develops myeloid malignancies, calling into question the potential clinical value of such wide-scale screening approaches. In fact, individuals older than age 80 years seem to have a normal life expectancy despite harboring aforementioned aberrations.148 Furthermore, Young et al demonstrated by using a more sensitive HTS approach that clinically silent clonal hematopoiesis can be detected in nearly all individuals between ages 50 and 60 (95%) and that malignant transformation in these cases is exceptionally rare.149 Those conflicting results highlight the need for future prospective longitudinal studies incorporating genetic tests to accurately define the risk of precancerous genomic conditions, to understand their clinical relevance, and to distinguish between benign and malignant clonal hematopoiesis. Consensus and guidelines for how to use detection of mutations in healthy individuals are required to inform clinical decision-making.
At AML diagnosis, profiling of certain gene rearrangements (eg, PML-RARA, CBFB-MYH11, RUNX1-RUNX1T1, BRC-ABL1) and mutations (eg, NPM1, FLT3-ITD, RUNX1, CEBPA) in BM or PB has become increasingly relevant for patient risk assessment and therapeutic decision-making.150-155 In addition, achieving deep CR without MRD is of key importance for assessment of clinical outcomes.155-157 MFC- and PCR-based technologies allow a highly sensitive (0.1%-0.001%), objective, and standardized assessment of treatment response over time (Figure 2A; Table 3). They provide independent prognostic information and are therefore recommended in trials and clinical practice for MRD detection.155-161 However, detection limits of those assays differ substantially between molecular markers, mainly because of large variations in expression levels of fusion genes and mutant alleles analyzed.155,162 Furthermore, it remains unclear what the best time to test for MRD might be; kinetics of MRD response differ considerably depending on treatment, methods, and markers tested (for a detailed review, see Döhner et al155 ).
MRD detection by HTS in AML/MDS is a relatively new field. Thol et al used amplicon HTS to follow FLT3-ITD and NPM1 mutations in BM and PB samples of a small cohort of AML patients. They demonstrated high concordance with qRT-PCR and robust detection of MRD, including the identification of emergent mutations during relapse.163 Kohlmann et al performed amplicon sequencing of RUNX1 in a cohort of 103 AML patients (both BM and PB samples), demonstrating that reduction of RUNX1 mutant alleles below a threshold predicts clinical outcome (Table 2).164 Most recently, Yeh et al showed that ctDNA analysis by targeted HTS accurately mirrors diagnostic BM genotypes, reflects clonal evolution over time, and predicts treatment failure in patients with MDS, indicating a potential future role for noninvasive disease monitoring.165
However, most of these studies either restricted their analyses to a subset of AML/MDS patients known to harbor one specific aberration, or applied HTS approaches with a relatively high lower limit of detection, allowing robust mutation detection only at AFs of 1% to 2%. Technical improvements will be needed to demonstrate a significant prognostic effect of HTS-based methods in large and genetically diverse patient cohorts.
In a screening study of 59 individuals from 17 families with familial AML/MDS, Churpek et al were able to identify recurrent pathogenic germ line alleles in 30% of cases by targeted HTS, including variants in GATA2 and RUNX1. Furthermore, the authors found that the spectrum of emerging somatic alterations in individuals with overt AML/MDS seems to be different from de novo AML.166 This suggests that high-risk families could potentially benefit from HTS for both recognition of pathogenic germ line alleles in asymptomatic carriers and early detection of emerging somatic alterations in individuals developing AML/MDS.
Chronic myeloid leukemia
Chronic myeloid leukemia (CML) is generally defined by the reciprocal chromosomal translocation between chromosomes 22 and 9, which results in an oncogenic BCR-ABL1 gene fusion. BCR-ABL1 is the target of highly effective tyrosine kinase inhibitors (TKIs; eg, imatinib) with excellent response rates.167 Monitoring of BCR-ABL1 transcripts under TKI treatment at certain disease milestones (“major molecular response” [MMR]) is greatly relevant for prediction of clinical outcomes.168-170
Established clinical tests to detect BCR-ABL1 and assess MMR include conventional cytogenetics, fluorescence in situ hybridization, and qRT-PCR.171 While cytogenetics is relatively insensitive and limited to cultured dividing BM cells, fluorescence in situ hybridization and qRT-PCR can be performed using noninvasively acquired PB cells. Moreover, qRT-PCR with a limit of detection of ∼0.001% also allows for the identification of “deep molecular remissions,” which may define a subgroup of patients who stay in unmaintained remission after treatment discontinuation.168,171-174 HTS-based approaches capturing BCR-ABL1 currently play only a minor role for response assessment in CML. Once fully established, they could facilitate MRD detection with even higher sensitivities. For example, Alikian et al used a coupled HTS and dPCR assay for detection of BCR-ABL1 breakpoints and demonstrated that this method outperforms qRT-PCR and DNA-based qPCR for MRD monitoring at deep molecular remissions.175
Resistance mutations scattered across the BCR-ABL1 kinase domain under TKI treatment occur frequently and are associated with disease progression, particularly in patients with rising BCR-ABL1 levels.176-178 Sanger sequencing is presently the primary method for identifying TKI resistance, but remains limited because of poor sensitivity (∼10%).179,180 HTS methods covering all known resistance mutations might overcome these limitations and facilitate prediction of TKI resistance and clinical progression. Soverini et al demonstrated that amplicon HTS allows detection of BCR-ABL1 kinase domain mutations down to 1% AF.181 In a study by Machova Polakova et al, amplicon-based HTS detected emergent TKI-resistant mutations in CTCs earlier than conventional sequencing, even at the time of MMR.180 Increasingly refined methods including duplex sequencing are likely to further improve detection and monitoring of such resistance mechanisms.182
Myeloproliferative neoplasms
BCR-ABL1 negative myeloproliferative neoplasms (MPNs) are characterized by BM proliferation of 1 or more myeloid lineages with no alterations in cellular maturation. Primary disease entities are polycythemia vera (PV), essential thrombocythemia (ET), and primary myelofibrosis (PMF).183 At molecular level, mutations in JAK2 V617F are frequently observed, with ∼95% of PV and 50% to 60% of ET/PMF patients harboring this aberration. However, variants in other genetic regions are also common, including CALR exon 9 (∼25% ET, ∼35% PMF), MPL exon 10 (∼4% PMF, ∼1% ET), and JAK2 exon 12 (∼4% PV).184-188
All those aberrations are part of diagnostic criteria in MPN and represent attractive targets for MRD monitoring.189 For tracking of JAK2 V617F, qPCR currently represents the most reliable and sensitive method (∼0.01%) and is widely used in clinical trials and routine laboratories to assess patient outcomes.190,191 For example, patients with JAK2 V617F levels >1% in PB or BM 1 month after allo-SCT appear to have a significantly higher risk of relapse and death than patients with deeper responses.191
Aberrations in JAK2 exon 12, MPL, and CALR are less established as markers for disease monitoring. However, a variety of different methods have been developed to track mutations in those genes, including qPCR and digital droplet PCR, achieving sensitivities of 0.01% to 2.5%.192-194 Some assays have already been used in clinical settings, demonstrating potential future utility for noninvasive disease monitoring.183,195-198
HTS-based approaches are not broadly established yet for routine MPN molecular diagnostics, but their capacity to simultaneously cover multiple genetic variants make them a promising tool for monitoring the genetic complexity of MPN.184,185,197 Abdelhamid et al demonstrated in a proof-of-principle study robust detection of JAK2 V617F in PB by HTS with high concordance to qPCR, even at low AFs.199 Lundberg et al applied a targeted HTS panel (104 genes) to serial blood samples and showed that the number of emergent mutations over time was low, characterizing these diseases as genetically stable. This study also illustrated the capability of HTS to detect the expansion of preexisting clones harboring TP53 and TET2 mutations toward transformation to AML.185 Similarly, Ferrer-Marín et al found the emergence of an ASXL1 mutation in a patient with JAK2 V617F–positive PMF, leading to leukemic transformation.200 A study performed by Fu et al suggested that identification of mutations in PB after allo-SCT by amplicon-based HTS results in a higher rate of MPN relapses.201
Because of the genetic complexity of MPN, we anticipate a growing role of HTS platforms in the future, both for mutation identification at diagnosis and disease monitoring.
Conclusions
Modern HTS approaches for noninvasive disease detection are not part of standardized procedures in hematologic malignancies yet. However, they have several decisive advantages over conventional approaches such as MFC and PCR-based assays, at least for selected applications (Figure 2; Table 3). First, targeted capture or amplicon HTS approaches are not restricted to one or only a few genetic markers and can capture hundreds or even thousands of genomic regions without sacrificing sensitivity. Second, the ability to detect and monitor multiple aberrations at the same time allows assessment of intra- and interpatient tumor heterogeneity, which facilitates the identification of clinically relevant subclone evolution. Finally, HTS approaches do not require individual and mutation-specific optimization or a priori knowledge of the patient’s tumor genotype.
Given the momentum of these advances, we envision that noninvasive HTS approaches will soon be shown to have clinical utility for precision and personalized therapeutic strategies in several hematologic cancers. For example, we anticipate a growing role in ALL, in which qPCR assays of clonal Ig/TCR gene rearrangements are currently considered the gold standard for assessment of MRD and are already included in clinical guidelines, despite several limitations. It has been demonstrated in numerous studies that HTS assays might overcome those limitations and thus have the potential to replace conventional methods as gold standard for MRD detection.57 Furthermore, we expect ctDNA analysis will soon be tested in trials for its clinical use in DLBCL and FL. Both neoplasms primarily manifest in lymph node tissue, hampering repeated sampling of tumor DNA. Applying IgHTS and targeted HTS to plasma samples at different disease milestones has demonstrated great potential for noninvasive risk assessment in several studies. Last, the majority of myeloid malignancies are characterized by a complex genetic landscape (eg, AML, MPN). Conventional PCR-based methods are highly limited by the proportion of patients they are able to cover. HTS platforms capturing the whole variety of genetic aberrations have particular advantages in these diseases and are expected to become part of patient management in the near future.
However, before HTS technologies can be implemented into routine clinical testing, certain challenges and limitations have to be addressed. For example, assays must undergo careful validation in multiple prospective clinical trials and prove that patients significantly benefit from their use. Moreover, disease-specific standardized workflows need to be defined by consortia bringing together specialists with clinical and technical experience and expertise. This must include a concerted effort to standardize the way samples are collected and quantified, methods for quality control of sequencing libraries, bioinformatics platforms, and data interpretation.
Authorship
Contribution: F.S. and D.M.K. wrote the manuscript and designed tables and figures; M.D. and A.A.A. revised the manuscript; and all authors reviewed and edited the final draft of the manuscript and made substantial contribution to discussion of the content.
Conflict-of-interest disclosure: M.D. and A.A.A. are coinventors on patent applications related to Cancer Personalized Profiling by Deep Sequencing. and are consultants for Roche Molecular Systems. The remaining authors declare no competing financial interests.
The current affiliation for F.S. is Department of Hematology, Oncology, and Stem Cell Transplantation, Freiburg University Medical Center, Albert-Ludwigs University, Freiburg, Germany.
Correspondence: Ash A. Alizadeh, Division of Oncology and Division of Hematology, Department of Medicine, Stanford University School of Medicine, Lorry Lokey Building, SIM 1, 265 Campus Dr, Stanford, CA 94305-5458; e-mail: arasha@stanford.edu.
