

Abstract
Advances in genetic labeling and barcoding of hematopoietic stem cells (HSCs) in situ now allow direct measurements of physiological HSC output, both quantitatively and qualitatively. Turning on a heritable label in HSCs and measuring the kinetics of label emergence in downstream compartments reveal rates of differentiation and self-renewal of HSCs and progenitor cells, whereas endogenous HSC barcoding probes physiological precursor-product relationships. Labels have been inserted at different stages of the hematopoietic differentiation hierarchy. Recent genetic and functional evidence suggests a phenotype (Tie2+) for tip HSCs. Fate mapping shows that many tip HSCs regularly feed into downstream stages, with individual cells contributing infrequently. Stem and progenitor cells downstream of tip HSCs serve as a major, nearly self-renewing source of day-to-day hematopoiesis, rendering the blood and immune system HSC-independent for extended periods of time. HSCs realize multilineage output, yet, fates restricted to several lineages or even a single lineage have also been observed. Single HSCs within a clone in the bone marrow that develop from a fetal HSC precursor have been observed to express clone-specific fates. Thus, the new tools probing HSC differentiation in situ are progressing beyond assays for HSC activity based on proliferation measurements and fates of transplanted stem cells, and the data challenge lineage interpretations of single-cell gene expression snapshots. Linking in vivo fate analyses to gene expression and other molecular determinants of cell fate will aid in unraveling the mechanisms of lineage commitment and the architecture of physiological hematopoiesis.
Approaching the physiology of hematopoiesis by precursor-product analyses
Hematopoietic stem cells (HSCs) give rise to a great variety of blood and immune cell lineages. Since the 1960s, the study of HSC differentiation has relied on in vitro colony assays and the transplantation of hematopoietic stem and progenitor cells (HSPCs) into myeloablated host animals; both sets of tools have reached considerable sophistication in probing the differentiation of single cells (eg, Yamamoto et al, Perié et al, Hoppe et al, Notta et al, Carrelha et al1-5 ). Recently, a new generation of experimental tools has been introduced that allows analyzing HSC output in situ (reviewed in Höfer et al6 ). Using these approaches, Sun et al7 and we8 have observed that physiological hematopoiesis and the reconstitution of the hematopoietic system after transplantation differ fundamentally with respect to HSC involvement and cell differentiation rates (reviewed in Busch and Rodewald9 ). These and subsequent studies5,10-12 on physiological precursor-product relationships of HSCs and their output now pave the way to use in vivo differentiation parameters as the basis for understanding the function of the hematopoietic system. At the same time, these emerging insights also show how much remains enigmatic.
The first group of open problems concerns long-standing questions on HSC self-renewal, differentiation, and loss. A few percent of HSCs are in the cell cycle on any given day.13-16 At least in theory, the resulting daughter cells can either serve to maintain the HSC compartment (self-renewal), and hence to compensate for cell loss, or differentiate and hence feed into progenitor pathways. In addition, HSCs might directly undergo differentiation without cell division (reviewed in Höfer et al6 ). The HSC compartment itself is heterogeneous, and the HSC subpopulation with the lowest proliferation rates, or with the longest label retention,13,14,17,18 has been interpreted to represent a “dormant” compartment from which stem cells can be called on demand into an “active” HSC compartment and subsequently may return to dormancy19 (reviewed in Trumpp et al20 ). How these proliferation-based HSC categories relate to HSC output in steady state or under stress remains to be elucidated.
Time-resolved fate mapping of physiological hematopoiesis measured the flux of differentiating cells generated from HSCs in vivo.8,10 In our study, we analyzed the output of Tie2+ HSCs, where recent evidence suggests that these are tip HSCs.8,21 Many of these tip HSCs regularly feed into downstream stages, with individual stem cells contributing infrequently. At the same time, stem and progenitor cells downstream of HSCs are major contributors to day-to-day hematopoiesis; these include short-term (ST)-HSCs and multipotent progenitors (MPPs), which, in steady state, exhibit considerable self-renewal,7,8,10 rendering the system for extended periods of times HSC independent.
The second group of unresolved questions concerns the structure of the hematopoietic system: the physiological lineage fates emerging from HSCs, and, closely related, developmental pathways linking HSCs, progenitors, and mature immune and blood cells. Interpretations of HSC fates have been based on various experimental systems that differ substantially from one another. Single HSC transplantation can address HSC properties after engraftment in irradiated recipients.1,2,5,22-24 Messenger RNA expression has been studied in mice and humans at the level of single HSPCs isolated ex vivo (eg, Moignard and Göttgens, Velten et al, Karamitros et al, Paul et al, Olsson et al,25-29 reviewed in Moignard and Göttgens and Papalexi et al25,30 ); for a comprehensive review of recent developments in HSC biology, see Laurenti and Göttgens.31 New approaches employing fate mapping and barcoding of native hematopoiesis5,7,8,10-12 now offer possibilities to reveal precursor-product relationships in physiological differentiation pathways.
A dynamic framework for physiological hematopoiesis
For several decades, the activity of HSCs in vivo has been characterized through their proliferative behavior. It had been recognized early on that HSCs have a lower division rate than downstream progenitor cells.32 To characterize their slow progression through the cell cycle, the term G0 state, or quiescent state, has become widely used. This G0 state is thought to correspond to a reversible exit from the cell cycle before cells cross the G1 restriction point; it must therefore be distinguished from the irreversible cell-cycle arrest in G1 in terminally differentiated cells such as in neurons or in senescent cells.33 Fate mapping approaches in tissues with high cell turnover, such as gut epithelium or hair follicles, have shown that rapidly proliferating and quiescent tissue stem cell populations may coexist.34,35 These findings have been interpreted in terms of a “division of labor,” with proliferating stem cells maintaining the tissue and the quiescent population serving as a reserve for heightened demand. In a similar vein, HSCs have been suggested to contain an active compartment that drives hematopoiesis in steady state, and a dormant reserve that preserves long-term self-renewal and responds to stresses.20 This view has been developed based on the observation of proliferative heterogeneity of HSCs.13 However, proliferating HSCs may not be identical with differentiation-active HSCs that generate progenitor compartments. For example, rarely dividing HSCs may contribute to differentiation while more often dividing HSCs could primarily proliferate for self-renewal and replacement of HSCs lost by differentiation.36,37 Thus, proliferation does not report on HSC output.38
More recently, several groups have developed mouse models for fate mapping of endogenous HSCs during hematopoiesis in vivo.8,10,39,40 We generated a tamoxifen-inducible Tie2-Cre (Tie2MeriCreMer) mouse that allows the heritable fluorescent labeling of Tie2+ cells in a highly specific manner (ie, without labeling of progenitor cells downstream from HSCs)8 at different stages of ontogeny.41 We refer to this as Tie2-driven fate mapping (Figure 1A). Although expression of Tie2 in HSCs has long been known at the population level,42 and been suggested to regulate quiescence,43 only recent experiments functionally characterized Tie2+ HSCs in vivo. In Tie2MeriCreMer knock-in mice8 and in transgenic Tie2-Gfp reporter mice,21 on the order of only 1% and 4%, respectively, of HSCs were marked (we refer here to cells marked in these genetic ways as Tie2+ HSCs). Collectively, recent evidence places Tie2+ HSCs at the top of the hematopoietic hierarchy:
Tie2+ HSCs have the highest reported reconstitution potential. Single-cell IV transplantation showed that 68% of Tie2+ HSCs reconstituted mice at long term as opposed to 17% for Tie2− HSCs. Of note, both of these HSC subsets had the same Lineage (Lin)−Sca+Kit+CD34−CD150+CD48−CD135− phenotype21 ; hence, the only difference was isolation based on Tie2 expression. Remarkably, single Tie2+ HSCs reconstituted 6/6 mice when transferred directly into the bone cavity of recipient mice.21
Engrafted Tie2+, but not Tie2− HSCs, regenerated Tie2+ HSCs after transplantation. Hence, Tie2+ HSCs are upstream of Tie2− HSCs.21
Tie2+ HSCs, genetically marked and fate mapped early in adult life, generated all downstream states and were maintained in the bone marrow for the entire lifetime of the mouse, consistent with self-renewal and protection of this compartment.8
Symmetric division, consistent with self-renewal and compartment maintenance, has been assayed for Tie2+ and Tie2− HSCs in an in vivo paired daughter cell assay. Symmetric divisions were found in Tie2+ HSCs but not in Tie2− HSCs.21
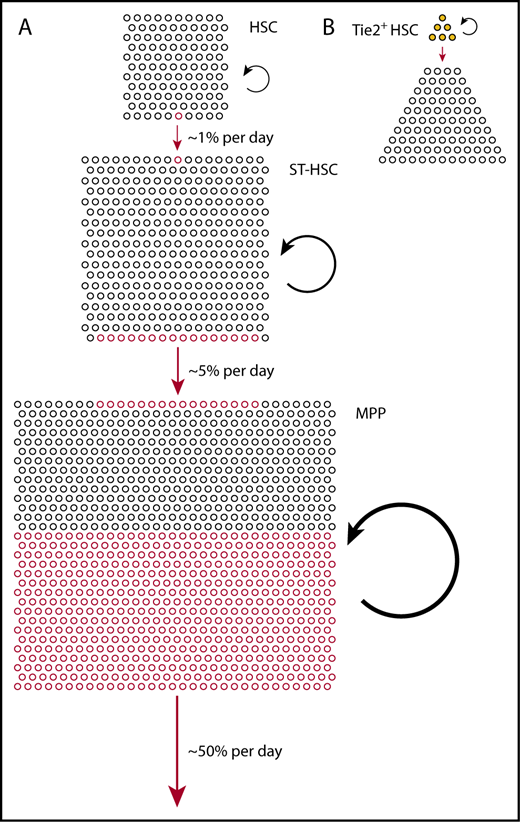
Physiological hematopoiesis is fueled by a hierarchically organized, broad basis of (almost) self-renewing stem and progenitor cells. (A) Relative compartment sizes of HSCs (Lin−Sca+Kit+CD150+CD48−), ST-HSCs (Lin−Sca+Kit+CD150−CD48−), and MPPs (Lin−Sca+Kit+CD150−CD48+) (all phenotypes according to Oguro et al15 ) are drawn to scale. Based on the measured output from Tie2+ HSCs,8 we estimated the frequencies of differentiating cells in each compartment, indicated by red circles (lower side, outgoing; upper side, incoming). The continuous but rare input from Tie2+ HSCs is greatly amplified in ST-HSCs and MPPs to sustain overall output (red arrows); to achieve this, the rates of self-renewing cell divisions also increase from HSCs to ST-HSCs to MPPs (black circle arrows). (B) Fate mapping data from Tie2MeriCreMer knock-in mice,8 and the comprehensive functional characterization of Tie2+ vs Tie2− HSCs in Tie2 reporter mice,21 indicate heterogeneity of the HSC compartment, with Tie2+ HSCs residing at the tip, differentiating, and self-renewing.
Physiological hematopoiesis is fueled by a hierarchically organized, broad basis of (almost) self-renewing stem and progenitor cells. (A) Relative compartment sizes of HSCs (Lin−Sca+Kit+CD150+CD48−), ST-HSCs (Lin−Sca+Kit+CD150−CD48−), and MPPs (Lin−Sca+Kit+CD150−CD48+) (all phenotypes according to Oguro et al15 ) are drawn to scale. Based on the measured output from Tie2+ HSCs,8 we estimated the frequencies of differentiating cells in each compartment, indicated by red circles (lower side, outgoing; upper side, incoming). The continuous but rare input from Tie2+ HSCs is greatly amplified in ST-HSCs and MPPs to sustain overall output (red arrows); to achieve this, the rates of self-renewing cell divisions also increase from HSCs to ST-HSCs to MPPs (black circle arrows). (B) Fate mapping data from Tie2MeriCreMer knock-in mice,8 and the comprehensive functional characterization of Tie2+ vs Tie2− HSCs in Tie2 reporter mice,21 indicate heterogeneity of the HSC compartment, with Tie2+ HSCs residing at the tip, differentiating, and self-renewing.
Hence, Tie2+ HSCs are located at the tip of the hematopoietic hierarchy, they fully self-renew and differentiate (Figure 1B).
These observations raise the question of the HSC output rate. In our experiments, labeling frequencies in compartments downstream of the Tie2–fate-mapped HSCs steadily approach the HSC labeling frequency in all mice but do so over very large timescales; even in the compartment immediately downstream of the HSCs, the ST-HSCs, the labeling frequency does not fully equilibrate over the lifetime of the mouse.8 Analysis of output rates show that an HSC gives rise to an ST-HSC on average once in 2 to 5 months (reviewed in Höfer and Rodewald38 ). Over the course of 9 months, >30% of Tie2+ HSCs produce differentiated progeny as determined by in vivo limiting dilution analysis.8 By contrast, the barcoding experiments using unique transposon integration sites suggest a lower contribution of HSCs to mature cells in adult mice (∼5%)7 ; the latter results may be affected by undersampling of mature cell clones and/or by the loss of barcodes by differentiation of HSCs out of the HSC pool.37
Our method of using time-resolved fate mapping to deduce HSC and progenitor cell output rates8 was subsequently applied by Sawai et al to data obtained in inducible Pdzk1ip1-CreER bacterial artificial chromosome transgenic mice.10 In this system, HSCs were labeled preferentially but not specifically (with 25% of the labeled Lin−Kit+ cells being Sca1− and hence having no stem cell/progenitor phenotype, and 33% of the labeled Lin−Kit+Sca1+ cells falling outside the HSC gate).10 The HSC labeling frequency increased until ∼ 1 year after label induction, which may be explained by heterogeneity within the HSC compartment.10 Using Pdzk1ip1-CreER for HSC fate mapping, Sawai et al estimated an HSC to give rise to an ST-HSC about once per 40 days, which indicates steady but again rather infrequent output, similar to our data.8 Nevertheless, this number is twice as high as the upper bound (95% confidence interval) of the HSC→ST-HSC output rate estimated with Tie2-driven fate mapping.8 Because Sawai et al10 do not provide model fits to the experimental data and accompanying statistics for their parameter estimates, potential causes for this quantitative discrepancy are difficult to analyze. In Sawai et al, broader labeling as well as the ad hoc assumption of “top-level” HSCs (without having phenotypic markers and hence actual data for this compartment) could both contribute. In a further recent fate mapping study, using keratin (Krt) Krt18-CreER45 or Fdg5-CreER,40 Chapple et al44 labeled not only HSCs but also cells further downstream from HSCs, notably MPPs and progenitors yet concluded that HSCs more actively contribute to adult hematopoiesis compared with previous studies.7,8,11,45,46 These discrepancies demonstrate that results from fate mapping experiments critically depend on the specificity of the Cre driver (ie, which stem and progenitor cell subpopulations are initially labeled by Cre). As soon as cells downstream from the cells under investigation are marked by Cre, it is impossible to link the origin of label emerging in the periphery to a defined source of cells in the bone marrow.
Slow HSC output rates estimated through time-resolved barcode dissemination7 and HSC fate mapping8 entail a remarkable prediction: If ST-HSCs receive little input from HSCs, then the major source of differentiated cells should be the ST-HSCs and downstream compartments. In turn, HSC ablation should not cause a significant shortage of mature blood and immune cells for many months.8 Indeed, Schoedel et al depleted HSCs by suicidal expression of diphtheria toxin to almost undetectable levels but recorded normal counts of white and red blood cells, platelets, and neutrophils in peripheral blood for nearly 7 months.45 Very similar results were reported independently by Sheikh et al after depleting HSCs by monocytic leukemia zinc-finger protein (KAT6A) knockout.46 Both of these papers provide a compelling experimental “mirror image” to the very slow HSC to ST-HSC output rate of once in 2 to 5 months observed with Tie2-driven fate mapping.8 Collectively, in our view, the unbiased transposon tagging throughout the hematopoietic system,7 the specific marking in combination with the Rosa-YFP reporter of tip HSCs in Tie2MeriCreMer knock-in mice,8 and the unbiased HSC ablation experiments45,46 are all consistent with low output rates from tip HSCs, and with largely HSC-independent maintenance of peripheral blood and immune cell compartments for extended periods of time under unperturbed conditions.
Given the low output of the HSC compartment, how are the enormous cell numbers needed to replenish comparatively short-lived hematopoietic cell populations, such as granulocytes, platelets, and erythrocytes, generated? The classical view places transit amplifying compartments in the differentiation hierarchy between stem cells and their mature products, implying that these compartments lack the self-renewal property characteristic of stem cells. The view that hematopoietic progenitors lack self-renewal was based on experiments in myeloablated mice, in which transplantation of donor stem or progenitor cells downstream from HSCs did not yield durable reconstitution of hematopoiesis (hence the name ST-HSC). However, the quantification of physiological cell differentiation and self-renewal rates in ST-HSCs and MPPs by fate mapping now indicates that these compartments are amplifying and, to a considerable extent, self-renewing (rather than transiting) in unperturbed hematopoiesis.8 As with stem cells, we define self-renewal here as the property of these progenitors to divide and yield new progenitors with the same functional properties as the parent cell; this process contributes to replacing cells lost by differentiation. Myeloid progenitors downstream of MPPs appear to lack this more extensive self-renewal.
In quantitative terms, ST-HSCs and MPPs amplify the cell flux by a factor of ∼1000 (ie, for each differentiating HSC in a given time interval, ∼1000 MPPs differentiate to downstream progenitor stages). This flux amplification is much larger than the cell number ratio for MPP/HSC: There are ∼10 MPPs per HSC but the MPPs also differentiate ∼100 times faster than the HSCs. Thus, by necessity, ST-HSC and MPP compartments can only be sustained if they have a high rate of self-renewing cell divisions (or else these compartments would be drained over time). Indeed, time-resolved fate mapping can be used to measure progenitor cell self-renewal in steady state because the rate at which labeled cells (stemming from labeled HSCs) enter a compartment is inversely proportional to the degree of self-renewal in the compartment.6,8 Of note, the fate mapping studies by us8 and Sawai et al10 agree, at least qualitatively, in yielding high rates of self-renewing divisions in ST-HSC and MPP in unperturbed hematopoiesis. In turn, this finding can explain the long persistence of cell clones that derive from transposon-tagged MPPs.7
In summary, the observation of HSC and progenitor cell output in unperturbed hematopoiesis by fate mapping supports a model of hematopoiesis in which the basis of (nearly) self-renewing stem cells is broadened beyond the HSCs and includes also the ST-HSC and, to a lesser extent, the MPP compartments. Many HSCs produce differentiated output in steady state, but do so with very low frequency; Tie2–fate-mapped HSCs at the tip of the hematopoietic hierarchy21 exhibit ∼ 1 differentiation event in a hundred cells per day.8 The output rate increases, with ∼5% of ST-HSCs and of the order of 50% of MPPs per day contributing to the respective downstream compartments in mice (Figure 1A). Consistent with increasing output frequencies and the need to maintain cell numbers in compartments, cell proliferation rates increase by similar orders of magnitude from HSC, to ST-HSC, to MPP.8,15 It will be interesting to study, upon challenges such as infection, bleeding, or irradiation (reviewed in King and Goodell47 ), the changes not only in proliferation but also in differentiation rates of stem and progenitor cells.
The concept of HSC dormancy and HSC fate mapping
The model that some HSC are dormant while others are active has mostly been based on findings of proliferative heterogeneity of HSCs, and differences in reconstitution potential in transplantation experiments depending on proliferative history of HSCs.13,17,19 Although the relationship between proliferation-based HSC categories and HSC output remains unclear, a perhaps tacit assumption has been that proliferation will generally be associated with output (leading to differentiation), whereas lack of proliferation (the dormancy hallmark) will be incompatible with differentiation. In theory, this would imply that, when selectively fate mapping dormant HSCs, only these should be marked, but cells downstream from dormant HSCs, such as progenitors and peripheral cells, would remain unlabeled at long term. By contrast, fate mapping of “active HSC” would result in equilibration between the label in HSC and the periphery. Currently, all fate mapping experiments reported HSC output, and hence, it remains unknown whether it will be possible to induce label in an HSC population that will not participate in hematopoiesis at long term. Although dormant HSCs have been proposed to be atop the hematopoietic hierarchy,19 the best characterized HSC population with tip properties are Tie2+ HSCs (see “A dynamic framework for physiological hematopoiesis”). Tie2+ HSCs have been fate mapped and, as a population, reliably and continuously produce differentiated output.8 It may be argued that the infrequent differentiation of individual Tie2+ HSCs (at a rate of once per 2-5 months) qualifies them as “dormant”; however, we believe that qualitative categories of this kind should eventually give way to a quantitative characterization of output rates.
Fates and developmental potential of HSCs
Fates, most notably multipotency, have been at the heart of the conceptual definition of HSCs from the very beginning. This notion dates back to the discovery of bone marrow cells with spleen colony-forming unit activity upon transplantation into lethally irradiated mice. Single-cell–derived colonies, or clones, often contained erythroid, granulocytic, and megakaryocytic cells. A single cell giving rise to progeny of more than a single lineage was consistent with a multipotent stem/progenitor cell, and the ability of some cells within these spleen colonies to form secondary colonies in further transplantation was in agreement with self-renewing stem cells.48,49 Aided by phenotypic enrichment of HSCs,50-52 the reconstitution of the entire blood and immune system of the mouse by transplantation of a single HSC became testable and revealed HSC potency in terms of engraftment, durable output, and multilineage in vivo reconstitution.23,24,51,53 Within this transplant setting, reconstituting HSC clones with either multipotent fates or fates restricted to a subset of lineages have reproducibly been observed.5,24,51,53,54 Such lineage patterns of reconstitution were often, but not always, stable across serial transplantations,5,23,54 suggesting some form of deterministic fates in HSC clones.55
Until recently, it was unknown to what extent the fates observed in posttransplantation hematopoiesis reflect normal HSC fates. Fates emerging from HSCs after engraftment of myeloablated recipients may be influenced by the vital need to rapidly replenish the empty system, and in this situation, most progeny are derived from small numbers of most successful donor HSC clones.56,57 Fates emerging from HSCs under normal conditions in vivo have recently been addressed by the development of nondisruptive methods to introduce large numbers (hundreds to thousands) of distinct genetic tags (barcodes) into HSCs in situ.11,12 Sun et al developed a transposon-based system that allows the inducible introduction of single-cell–marking transposon integration sites into essentially all hematopoietic cells, including, but not being selective for, stem or progenitor cells.7 We developed a Cre-recombinase–driven endogenous barcoding system, termed Polylox, that generates highly diverse genetic barcodes by DNA recombination in Rosa26Polylox reporter mice11 (for an illustration, see Figure 2). In this system, the use of inducible Cre lines can generate barcodes in a tissue- and time-controlled manner. Both of these endogenous barcoding systems have been used to address fates emerging from HSCs in situ,11,12 the former by exploiting the normal turnover of progenitor cells.
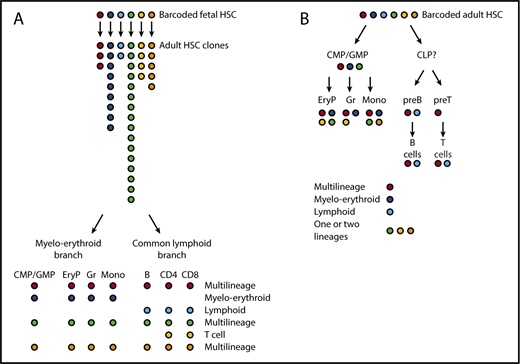
The tracing of HSC barcodes introduced in situ supports a treelike model of hematopoiesis with major myeloerythroid and common lymphoid branches. (A) We introduced barcodes during fetal development in the mouse (embryonic day 9.5). Labeled clones of adult HSC (analyzed at ∼1 year of age) are of very different sizes. HSC clones typically yield multilineage progeny, or oligolineage progeny either on the myeloerythroid or on the lymphoid side. (B) When barcoding HSCs in adult mice, HSCs again give rise to multilineage or oligolineage (typically, myeloerythroid or lymphoid) progeny. In addition, HSC-derived barcodes are also found in more restricted sets of mature cells (bi- or unilineage). Overall, HSC-derived barcodes co-occur in mature lineages and in their respective progenitors. For example, barcodes enriched in erythroid progenitors (EryP), granulocytes (Gr), or monocytes (Mono) are likely to be enriched also in CMPs/GMPs, whereas barcodes prevalent in mature T and B cells are also prevalent in the respective progenitors but not in CMPs. Colors symbolize the genetic Polylox barcodes on which this figure is based.11 EryP, erythrocyte progenitor; preB, precursor B cell; preT, precursor T cell.
The tracing of HSC barcodes introduced in situ supports a treelike model of hematopoiesis with major myeloerythroid and common lymphoid branches. (A) We introduced barcodes during fetal development in the mouse (embryonic day 9.5). Labeled clones of adult HSC (analyzed at ∼1 year of age) are of very different sizes. HSC clones typically yield multilineage progeny, or oligolineage progeny either on the myeloerythroid or on the lymphoid side. (B) When barcoding HSCs in adult mice, HSCs again give rise to multilineage or oligolineage (typically, myeloerythroid or lymphoid) progeny. In addition, HSC-derived barcodes are also found in more restricted sets of mature cells (bi- or unilineage). Overall, HSC-derived barcodes co-occur in mature lineages and in their respective progenitors. For example, barcodes enriched in erythroid progenitors (EryP), granulocytes (Gr), or monocytes (Mono) are likely to be enriched also in CMPs/GMPs, whereas barcodes prevalent in mature T and B cells are also prevalent in the respective progenitors but not in CMPs. Colors symbolize the genetic Polylox barcodes on which this figure is based.11 EryP, erythrocyte progenitor; preB, precursor B cell; preT, precursor T cell.
In their experiments measuring the propagation of tags along hematopoiesis, both groups found high proportions of barcodes shared between myeloid and erythroid lineages, consistent with a common pathway toward granulocytes, monocytes, and erythrocytes. The Polylox system also revealed close barcode links between T and B cells, largely distinct from myeloerythroid development. As a caveat, the different lifespans of cells may impact the sampling of barcodes at given time points. The same holds true for analyzing limited samples in which barcodes may be missed. Nevertheless, these in vivo barcode data are consistent with normal hematopoiesis exhibiting distinct myeloerythroid and common lymphoid pathways emanating from HSCs. Although this treelike structure had been originally proposed mostly based on the identification of transplantable common lymphoid progenitors (CLPs),58 common myeloid progenitors (CMPs), granulocyte monocyte progenitors (GMPs), and megakaryocyte erythrocyte progenitors (MEPs),59 much controversy has surrounded this model (eg, Notta et al, Velten et al, Kawamoto et al4,26,60 see also “Heterogeneity of hematopoietic progenitor cells”). In our view, the key criterion for the appraisal of hematopoietic differentiation models is the measurement of precursor-product relationships, under conditions as close as possible to normal physiology. Thus far, the data obtained by in vivo fate mapping of native hematopoiesis11,12 support 2 major (ie, myeloerythroid and lymphoid) branches. With the possible exception of megakaryocytes (discussed in “Heterogeneity of hematopoietic progenitor cells”), these branches are compatible with the original model of hematopoiesis that was largely derived from transplantation of HSPCs.
Did the in vivo barcoding experiments reveal evidence for multipotency of HSCs in vivo? We found common barcodes in myeloid, erythroid, and lymphoid cells that arose from stem cells in 2 settings: from barcoded embryonic HSC that gave rise to adult HSC clones (Figure 2A), and from single barcoded HSPCs in adult hematopoiesis (Figure 2B).11 Independent support for the existence of multipotent HSCs was provided by Rodriguez-Fraticelli et al who found, increasingly with time, codes shared between myeloid, erythroid, and B-cell lineages (T cells were not sampled).12 These collective data demonstrate multilineage output for normal HSCs in the bone marrow in situ, during both development and adult maintenance of the hematopoietic system. However, it appears that not all HSCs use multipotent fates because both Polylox and transposon barcoding also revealed lineage-restricted outputs, including only erythroid-myeloid or only lymphoid (and other combinations), and, especially in adult steady-state hematopoiesis, single-lineage fates.11,12 As a caveat for the interpretation of these data, we note that the rare output of adult HSCs8 could make it more difficult to detect multilineage outcomes in limited samples of mature cells. Nevertheless, there is evidence for lineage-restricted fates.
When barcodes were induced at embryonic day 9.5, a time when HSCs arise,61 lineage-restricted fates from adult HSC clones, derived from single HSC progenitors, suggest that cells can have clone-intrinsic or coherent fate restriction. This interpretation is in line with a study that achieved endogenous color-coding of HSCs to follow the fates emerging from labeled HSC clones in situ and after transplantation.54 Interestingly, labeled clones showed over time, and after transplantation, stereotypical fate behaviors, which may be controlled by DNA methylation.54 As it seems unlikely that individual cells belonging to an HSC clone reside as a cluster in one and the same local niche neighborhood,62 the niche context may have little instructive role in driving HSC fates.63
Heterogeneity of hematopoietic progenitor cells
A phenotype for a CMP had originally been identified based on colony assays and transplantation.59 Subsequent work using these traditional assays and refined progenitor cell phenotypes,64,65 expression of lineage-specifying transcription factors,66 or progenitor barcoding2 have found considerable fate heterogeneity within the CMP compartment. Nevertheless, all these studies detect at least a CMP subpopulation that gives rise to both myeloid and erythroid fates at a clonal level. In particular, erythroid and diverse myeloid progenies originate from single barcoded progenitors robustly in hematopoietic differentiation posttransplantation.2 However, this oligolineage outcome is enriched within the MPP compartment compared with the phenotypic CMPs. Hence, Perié et al proposed that the origin of the common myeloid progenitors is mainly in the MPPs.2
The existence of such oligolineage progenitors has become subject to controversy.1,4,5,12,26,28 As an approach to study physiological hematopoiesis, several groups have turned to single-cell transcriptome analyses. Paul et al have described pervasive heterogeneity of gene expression within the CMPs28 and suggested that the CMP compartment decomposes into distinct cell clusters that are “transcriptionally primed” toward single differentiation fates, and no progenitors with a mixed state exist in the CMPs. A recent single-cell transcriptome study of human hematopoiesis goes further by suggesting that lineage-restricted oligo- or bipotent progenitor cells do not occur at all in the hematopoietic system.26 This view would imply fundamental differences between physiological and posttransplantation hematopoiesis or/and the functioning of the system in humans vs mice, as single functional CMPs are transplantable in mice.2 Moreover, in the experiments by Perié et al,2 functional CMPs with an MPP phenotype are the dominant producers of both erythrocytes and myeloid cells, suggesting that the main differentiation pathways for both kinds of cells pass through a common intermediate stage.
More recent studies have interpreted single-cell transcriptome data of the same cell populations to include oligo- and bipotent progenitor cells. In contrast to Paul et al,28 Olsson et al29 suggest the existence of mixed bipotential states in myeloid progenitors from the mouse, which can give rise to both neutrophil and monocyte fates. Interestingly, reanalysis67 of the myeloid progenitor data from Paul et al28 with a method that stresses continuity (diffusion pseudotime67 ) suggests a small population of common myeloid progenitors with a downstream split into a megakaryocyte-erythrocyte lineage branch and a granulocyte-macrophage branch, in agreement with the classical model and its subsequent refinements.2,59,64,66 Unlike Velten et al,26 Karamitros et al27 suggest the existence of bi- and oligopotent progenitors for human hematopoiesis. In both Olsson et al29 and Karamitros et al,27 bi- and oligopotent progenitor phenotypes are rare in the cell populations analyzed. However, low frequency does not argue against such states as common intermediates in physiological differentiation pathways in vivo. Of note, a recent interrogation of the gene-regulatory landscape of human hematopoiesis by single-cell ATAC-seq (assay for transposase-accessible chromatin using sequencing) provided evidence for common progenitors of myeloid and erythroid lineages.68
Nondisruptive fate mapping may help to interrogate this question further. Although single-cell transcriptomes give an unprecedented view of gene expression heterogeneity, they are static snapshots and hence do not provide precursor-product relationships. To link cell transcriptomes with development, the notion of “transcriptional priming” has been put forward.26,28 According to this idea, the expression of lineage-specifying transcription factors or other lineage-specific genes by stem or progenitor cells should reflect their commitment to a particular lineage. Clearly, lineage-committed progenitors express specific regulators; however, also uncommitted HSCs can express such genes without compromising their multipotency. In a marked example, 50% of singly transplanted von Willebrand-factor (Vwf)- (a megakaryocyte hallmark) expressing and Gata1 (an erythrocyte hallmark) coexpressing HSCs yield fully multipotent (erythroid, megakaryocyte, myeloid, and B and T cell) output; moreover, fate mapping in Vwf-CreERT2 mice includes nonmegakaryocytic lineages.5 Further examples of lineage gene expression prior to commitment, indeed contradicting the final fate, are IL7R-Cre mice69 in which a small fraction of HSCs are marked yet not lymphocyte-committed, or LysM-Cre mice in which prior to macrophage specification >5% of all hematopoietic stem and progenitors are marked, with no consequence for their future fates.70 Also, HSCs express PU.1 and depend on this transcription factor for their maintenance, although further downstream in the differentiation hierarchy PU.1 functions as a hallmark myeloid factor.71 In particular, PU.1 protein expression in single stem cells does not preclude the read-out erythroid fate.3 These cases show that the question of how early lineage commitment can be inferred from lineage gene expression remains poorly understood.
To our knowledge, the fate of individual progenitor cells has not yet been mapped under physiological conditions in vivo. Nevertheless, 2 recent studies support the existence of myeloerythroid progenitors in physiological hematopoiesis.11,12 Rodriguez-Fraticelli et al labeled individual HSPCs by transposon integration and reported common myeloerythroid fates. After barcode induction, shared barcodes were found so early in monocytes, granulocytes, and erythroblasts that they likely originated from a common progenitor rather than from HSCs. After fetal HSC barcoding and measuring the output of adult HSC clones, or barcoding and output measurements from adult HSC, we observed very similar barcodes and barcode frequencies in erythroid progenitors and granulocytes in adult mice, suggesting a common developmental origin of both lineages.11 Further consistent with this idea, about two-thirds of these barcodes were also found in CMPs with very similar frequencies as in the downstream lineages. However, one-third of the shared erythroid progenitor and granulocyte barcodes were not recovered from the CMPs (although relative to the total CMP number, CMPs were sampled more comprehensively than erythroid progenitors and granulocytes). This may be due to very limited self-renewal of CMPs in the bone marrow,8 or point to additional differentiation pathways that “bypass” the phenotypic CMPs.
The classical model predicted that platelets arise from the MEP stage, downstream from CMPs,59 via phenotypically defined megakaryocyte-committed progenitors among erythromyeloid progenitors.64,65 By contrast, a potential shortcut from HSCs to megakaryocytes has been suggested under inflammatory conditions,72 and recent reports, based on barcoding,12 single cell transplantation, and Vwf-driven inducible fate mapping,5 may point at a megakaryocyte-restricted subset of long-term HSCs, or even a shortcut from HSC to megakaryocytes under steady-state conditions. However, some uncertainties remain. The assayed megakaryocyte progenitor phenotype (Lin−Kit+Sca1−CD150+CD41+)12 is, with the exception of Sca1 expression, identical to HSCs expressing CD41,1 and hence, HSCs losing or transiently downregulating Sca1 may fall into this gate. Common features of megakaryocytes and HSCs have been noticed.73 In addition to transplantation, Carrelha et al also used inducible Vwf-CreERT2 mice to track progeny of Vwf-expressing cells in situ.5 This experiment also labels, in addition to the fraction of (∼40%) Vwf-expressing HSC, Vwf-expressing megakaryocyte progenitors, which may be sufficiently long lived to first dominate lineage tracing of platelets, until, at later time points, output from multipotent Vwf-expressing HSCs takes over, a scenario that would fit the actually observed data.5 Lineage tracing experiments in Flk2/Flt3-Cre mice further argue against a direct shortcut from HSCs to megakaryocytes, where the vast majority of platelets arise from an Flk2/Flt3-Cre expressing intermediate stage under steady-state conditions, after irradiation-induced stress and upon HSC transplantation.74 Another study also found strong megakaryocyte lineage labeling in Flk2/Flt3-Cre mice, but, in contrast to Boyer et al,74 here the labeling also included ∼23% of HSCs.75 Collectively, further work will be required to clarify the route(s) from HSCs to platelets, including their quantitative contributions.
Acknowledgments
The authors thank Katrin Busch, Ann-Katrin Schuon, Kay Klapproth, Melania Barile, Weike Pei, Thorsten Feyerabend, and Jens Rössler as well as other members of the Rodewald and Höfer laboratories for experiments leading to the described concepts and for ongoing discussions, and Katrin Busch for comments on the manuscript.
This work is supported by Sonderforschungsbereich (SFB) 873-B11 and European Union project 764698-QuanTII (T.H., H.-R.R.) and European Research Council Advanced grant 742883 (H.-R.R.).
Authorship
Contribution: T.H. and H.-R.R. wrote this perspective.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Thomas Höfer, German Cancer Research Center, Im Neuenheimer Feld 280, 69120 Heidelberg, Germany; e-mail: t.hoefer@dkfz.de; and Hans-Reimer Rodewald, German Cancer Research Center, Im Neuenheimer Feld 280, 69120 Heidelberg, Germany; e-mail: hr.rodewald@dkfz.de.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal