

In this issue of Blood, have used a novel cell-based assay to evaluate the antagonist effects of 4 clinically prescribed vitamin K antagonists (VKAs) on the activity of vitamin K epoxide reductase (VKOR) and its VKA-resistant mutants in their natural cellular environment.1
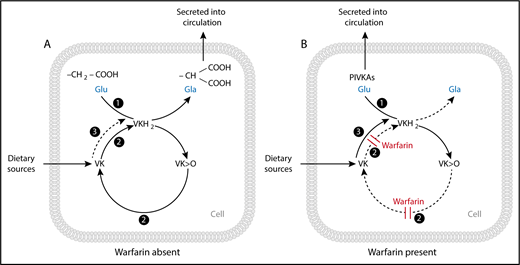
Enzyme activities of the vitamin K (VK) cycle in the (A) absence and (B) presence of warfarin. (A) The enzyme γ-glutamyl carboxylase (GGCX) (activity 1) with the cofactor vitamin K hydroquinone (VKH2) facilitates the transformation of peptide-bound glutamate (Glu) to γ-carboxyglutamate (Gla) residues and the subsequent synthesis and secretion of carboxylated VK-dependent proteins. The γ-carboxylation reaction results in the generation of VK epoxide (VK>O), which is reduced to VK quinone by the enzyme VK epoxide reductase (VKOR) (activity 2). VK quinone is then reduced to the VKH2 cofactor by 1 or more unidentified NAD(P)H-dependent reductases (activity 3), or possibly by VKOR itself (activity 2), to complete the cycle. (B) In the presence of a vitamin K antagonist (VKA) such as warfarin, VKOR (activity 2) is inhibited, resulting in the synthesis and secretion of inactive species of undercarboxylated proteins called proteins induced by vitamin K absence or antagonism (PIVKAs). Given sufficient input of vitamin K into the cycle, an alternative quinone reductase pathway (activity 3) can bypass the VKOR to provide the VKH2 substrate for GGCX and hence overcome the inhibitory action of warfarin, even under extreme blockade. Reproduced with permission from Shearer and Okano.3
Enzyme activities of the vitamin K (VK) cycle in the (A) absence and (B) presence of warfarin. (A) The enzyme γ-glutamyl carboxylase (GGCX) (activity 1) with the cofactor vitamin K hydroquinone (VKH2) facilitates the transformation of peptide-bound glutamate (Glu) to γ-carboxyglutamate (Gla) residues and the subsequent synthesis and secretion of carboxylated VK-dependent proteins. The γ-carboxylation reaction results in the generation of VK epoxide (VK>O), which is reduced to VK quinone by the enzyme VK epoxide reductase (VKOR) (activity 2). VK quinone is then reduced to the VKH2 cofactor by 1 or more unidentified NAD(P)H-dependent reductases (activity 3), or possibly by VKOR itself (activity 2), to complete the cycle. (B) In the presence of a vitamin K antagonist (VKA) such as warfarin, VKOR (activity 2) is inhibited, resulting in the synthesis and secretion of inactive species of undercarboxylated proteins called proteins induced by vitamin K absence or antagonism (PIVKAs). Given sufficient input of vitamin K into the cycle, an alternative quinone reductase pathway (activity 3) can bypass the VKOR to provide the VKH2 substrate for GGCX and hence overcome the inhibitory action of warfarin, even under extreme blockade. Reproduced with permission from Shearer and Okano.3
The first VKA to be used clinically was bishydroxycoumarin (dicumarol), the compound identified by Link’s group in Wisconsin as the anticoagulant present in spoiled sweet clover that was the cause of an often lethal hemorrhagic disease in cattle.2 Clinical trials of dicumarol as an oral anticoagulant began in 1940 and, within 2 years, more than 100 related 3-substituted, 4-hydroxycoumarins had been synthesized by Link’s laboratory for biochemical and clinical appraisal. One of these compounds was warfarin which was originally thought to be too potent for clinical use but which Link thought would make an ideal rodenticide.2 Indeed, after its introduction in 1948, warfarin revolutionized rodent control and, after a failed suicide attempt by a naval officer, equally revolutionized oral anticoagulant therapy.2 Today, warfarin is still the most commonly prescribed oral VKA, but some countries use the related 4-hydroxycoumarins acenocoumarol and phenprocoumon or the 1,3-indandione derivative fluindione. All these VKAs were introduced into clinical practice without much knowledge of their molecular mechanism of action. It was not until the 1970s that the enzyme VKOR was identified as the target enzyme which, when inhibited, blocks the hepatic synthesis of active (γ-carboxylated) vitamin K-dependent coagulation factors (see figure).3 Because of the indirect mechanism of VKA action, a single blocking dose of warfarin does not produce the maximum hypoprothrombinemic response until 2 to 4 days after administration.4 This delayed pharmacodynamic response together with variable pharmacokinetics makes it difficult to compare the in vivo potency of different VKAs in humans.
The 2 major enzymes of the vitamin K cycle, γ-glutamyl carboxylase (GGCX) and VKOR, are integral membrane proteins that reside in the endoplasmic reticulum of cells and require cofactor forms of vitamin K that are themselves hydrophobic. For VKOR, the difficulty in evaluating structure-function relationships is that traditional functional assays are carried out in detergent-solubilized microsomal fractions using the nonphysiological reductant dithiothreitol (DTT) as a substitute for the unknown physiological reductants. The inherent artificiality of DTT-driven assays often provides an inaccurate picture of VKOR activity in vivo. After using such assays, only about one-third of missense VKOR variants defined as resistant to first-generation VKAs in humans or rodents showed the expected degree of resistance.5-7
The study of GGCX and VKOR has been significantly advanced by the introduction of cell- based reporter assays in conjunction with gene-editing techniques.8 The principle is to express a chimeric vitamin K-dependent reporter protein in mammalian cells in which the reporter γ-carboxylation status can be readily measured by an enzyme-linked immunosorbent assay. In this way, one can knock out the specific gene of interest (GGCX or VKOR) in the reporter cell line and introduce a recombinant gene expressing the desired mutant in its place.8
The first objective of Chen et al was to make a direct comparison of the inhibitory potency of 4 clinical VKAs as measured by the efficiency of carboxylation of the chimeric reporter protein and the half-maximal inhibitory concentrations of each VKA. The standout finding was that the efficacy of inhibition of wild-type VKOR followed the order of acenocoumarol > phenprocoumon > warfarin > fluindione, with the efficacy of acenocoumarol being fivefold to eightfold higher than that of the other VKAs. These observations were then extended to the determination of the carboxylation efficiency in the same cell-based assay when the cells expressed each of the 27 clinically identified VKA-resistant mutations of VKOR. This substudy determined that most but not all of these VKOR mutations showed a degree of resistance consistent with the clinical picture. Acenocoumarol, which had already been shown to be the most potent inhibitor of VKOR-dependent carboxylation, showed the least variability of response among the 27 resistant variants.
Another question is whether the 27 VKA-resistant mutations of VKOR show diminished VKOR activity in the absence of VKAs, as had been found for several variants using the DTT-driven assay.5 Chen et al showed that all 27 variants were fully functional in their cell-based assay, which is consistent with findings of normal hemostasis in nonanticoagulated persons with these resistant variants. Interestingly, there was a good correlation among these variants between the binding of warfarin and vitamin K epoxide to VKOR, suggesting that the mutations affect how both the drug and substrate bind to the enzyme.
In summary, Chen et al present the first comprehensive quantitative comparison of the interaction of 4 common VKAs with VKOR and its VKA-resistant variants in a cellular model that closely mimics the membrane environment in which the vitamin K cycle operates. Not all clinically identified VKA-resistant VKOR mutations resulted in VKA-resistant VKOR activity, even in this cell-based system; this suggests that factors other than VKOR were responsible for their clinical resistance. It is unlikely that the data will, or should, cause physicians to change their current practice except perhaps (as the authors suggest) to include the relevant missense mutations in dosage algorithms. On the rare occasions that resistant mutations are encountered in the clinic, Chen et al present evidence that for some mutations, the degree of resistance differs widely between different VKAs. In such cases, it may be beneficial to switch to a VKA that in this model predicts a lower dose requirement for stable anticoagulation. Other relevant factors that the data do not take into account are the effects of VKA stereoisomerism and metabolism. For example, using a similar cell-based assay system, Haque et al9 showed differential VKOR inhibition by warfarin enantiomers (S>R) consistent with their in vivo potencies and that 10-hydroxywarfarin and warfarin alcohol metabolites show significant inhibition of VKOR. Apart from VKAs used clinically, second-generation VKAs that belong to the so-called superwarfarin family are powerful, long-acting rodenticides that pose significant health risks from accidental and nonaccidental poisoning.10 This should prove to be another fruitful area for the application of these novel cell-based assays.
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal