

In this issue of Blood, report a mechanism underlying susceptibility to Aspergillus fumigatus infections in patients treated with the Bruton tyrosine kinase (Btk) inhibitor ibrutinib.1 This drug has been paradigm-shifting in a number of B-cell malignancies, which has led to widespread use in the diseases for which Food and Drug Administration approval currently exists, and clinical trials of the drug alone and in combination in these and many other malignancies. However, although well tolerated in the majority of patients, a few troublesome toxicities have become apparent as use of the drug has expanded. One such toxicity is a relatively high rate of A fumigatus infections. Although patients with hematologic malignancies are certainly at risk for opportunistic infections, A fumigatus infections at a higher rate than expected have been noted in multiple prospective studies, most notably a rate of 39% in patients on a trial of ibrutinib in primary central nervous system (CNS) lymphoma who were concurrently treated with corticosteroids.2 Although the incidence in other hematologic malignancies is likely quite a bit lower (2.5% in a large institutional cohort of 566 patients treated over 1225 person-years),3 this finding has been worrisome. A previous study by this group, exploring the risk of A fumigatus in patients treated with calcineurin inhibitors, demonstrated that Btk is required for the macrophage inflammatory response that clears A fumigatus from the airways,4 and a separate study showed that Btk null mice infected with A fumigatus developed pneumonia, similar to what is seen in macrophage-deplete models.2 However, the implications for these findings in patients where Btk is present but pharmacologically inactivated were previously unknown.
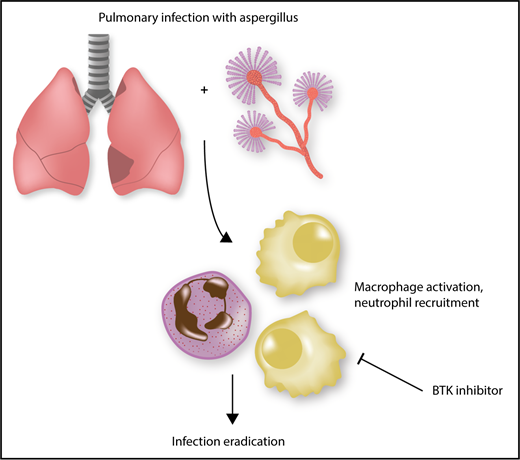
Eradication of A fumigatus is inhibited by ibrutinib. Effective clearance of Aspergillus requires macrophage activation and subsequent neutrophil recruitment. This activation is a Btk-dependent process, and inhibitors of Btk, such as ibrutinib, preclude effective clearance of A fumigatus, predisposing patients to invasive infection.
Eradication of A fumigatus is inhibited by ibrutinib. Effective clearance of Aspergillus requires macrophage activation and subsequent neutrophil recruitment. This activation is a Btk-dependent process, and inhibitors of Btk, such as ibrutinib, preclude effective clearance of A fumigatus, predisposing patients to invasive infection.
In this study, the authors show that the macrophage activation induced by A fumigatus exposure is inhibited by ibrutinib, and similarly, ibrutinib inhibits nuclear translocation of NFAT and NF-κB in these cells, which has previously been shown to be critical to neutrophil recruitment (see figure). Furthermore, human monocyte–derived macrophages (hMDMs) and alveolar macrophages are unable to produce tumor necrosis factor -α in response to A fumigatus after Btk knockdown, and galactomannan production is inhibited in ibrutinib-treated hMDMs. However, macrophage phagocytosis is not affected by ibrutinib.
This study is significant because it both points to a mechanism behind an important clinical observation and extends the knowledge of the complex interplay between ibrutinib and the innate immune microenvironment. Data so far on the effects of ibrutinib on innate immune cells have been quite mixed. Ibrutinib has been shown to limit natural killer cell antibody–dependent cellular cytotoxicity5 in vitro; however, other data suggest that when cocultured with monocytes, chronic lymphocytic leukemia (CLL) killing is not affected by ibrutinib.6 As well, there are data showing that ibrutinib promotes the activity of the CLL-supporting nurselike cells,7 but patient data have also shown that ibrutinib disrupts the interaction between macrophages and CLL cells in the bone marrow microenvironment.8 Also, there are data to suggest that ibrutinib impairs macrophage phagocytosis,9 but other data that show that monocyte phagocytosis is not affected.6 Overall, these data serve to highlight the complicated nature of the interplay between Btk and the tumor microenvironment and show the importance of continued efforts in this area and in the construct of in vitro conditions that mimic the complete microenvironment.
Clinically, the finding that A fumigatus infections in patients on ibrutinib are due to a specific defect in immunity induced by this drug is quite significant. Although the absolute risk is certainly low, it will be important to identify those patients at particularly high risk for infection. It appears from the CNS lymphoma trial that patients taking concomitant corticosteroids are at especially high risk. Other risk factors that have been identified include high number of prior therapies, diabetes, and liver disease.3 Unfortunately, antifungal prophylaxis is not simple with ibrutinib, because most antifungal agents inhibit CYP3A4 and thus raise levels of ibrutinib. Further work will be necessary to definitively determine which patients are likely to derive benefit from antifungal prophylaxis, and potentially which patients would be better served with alternative therapies. Although this risk certainly does not dampen enthusiasm for this effective drug, it serves as a reminder that no therapy is without risk, and continued work to develop the optimal agents and strategies for treating our patients with hematologic malignancies must remain a top priority.
Conflict-of-interest disclosure: J.A.W. receives research funding from Janssen, AbbVie, Acerta, Karyopharm, and Morphosys.
