

Abstract
Immunodeficiency-associated lymphoproliferative disorders (IA-LPDs) are pathologically and clinically heterogeneous. In many instances, similar features are shared by a spectrum of IA-LPDs in clinically diverse settings. However, the World Health Organization (WHO) classifies IA-LPDs by their immunodeficiency setting largely according to the paradigm of posttransplant lymphoproliferative disorders but with inconsistent terminology and disease definitions. The field currently lacks standardization and would greatly benefit from thinking across immunodeficiency categories by adopting a common working vocabulary to better understand these disorders and guide clinical management. We propose a 3-part unifying nomenclature that includes the name of the lesion, associated virus, and the specific immunodeficiency setting for all IA-LPDs. B-cell lymphoproliferative disorders (LPDs) are usually Epstein-Barr virus (EBV)+ and show a spectrum of lesions, including hyperplasias, polymorphic LPDs, aggressive lymphomas, and, rarely, indolent lymphomas. Human herpes virus 8–associated LPDs also include polyclonal and monoclonal proliferations. EBV− B-cell LPDs and T- and NK-cell LPDs are rare and less well characterized. Recognition of any immunodeficiency is important because it impacts the choice of treatment options. There is an urgent need for reappraisal of IA-LPDs because a common framework will facilitate meaningful biological insights and pave the way for future work in the field.
Introduction
Immunodeficiency-associated lymphoproliferative disorders (IA-LPDs) are a heterogeneous group of lesions with variable clinicopathologic features. The World Health Organization (WHO) classification recognizes 4 types of IA-LPDs: posttransplant lymphoproliferative disorders (PTLDs), lymphomas associated with HIV infection, lymphoproliferations associated with primary immune disorders, and other iatrogenic IA-LPDs.1 In the WHO classification, these IA-LPDs are described in 4 separate chapters according to the underlying clinical risk factors. This categorization is largely based on clinical knowledge and specific therapeutic options used in each of those settings. This current approach ignores common oncogenic, biological, and pathological features among various immunodeficiency settings and instead emphasizes the distinctive features that are characteristic of each setting. Despite shared histology, immunophenotype, and genetic features, the WHO classification arbitrarily separates IA-LPDs and leads to the use of different terminology, and sometimes even different diagnostic criteria, for similar IA-LPDs occurring in various immunodeficiency settings. Novel types of IA-LPDs that have emerged in the face of newer therapeutic agents are not mentioned in the current classification, and other less-recognized immunodeficiency settings, such as immune senescence, have not been included as causes of immunodeficiency.
Prompted by the need for reappraisal of the current approach to the diagnosis of IA-LPDs, the Society for Hematopathology and the European Association for Haematopathology conducted a workshop on immunodeficiency and dysregulation in October of 2015. In this perspective, we aim to provide a common framework for IA-LPDs that will allow a systematic approach for further study and support meaningful comparisons and interpretation of data, such that diagnostic criteria can be better defined. The adoption of a common framework with unified terminology that can be applied across clinical settings would be beneficial in deriving biological insights, predicting clinical behavior, and developing novel treatment strategies.
Proposed unifying framework for the classification of IA-LPDs
At the Society for Hematopathology and the European Association for Haematopathology workshop and in the corresponding proceedings,2-7 a shared working vocabulary was proposed based on a 3-part unifying nomenclature for all IA-LPDs: (1) the name of the lesion or the closest approximation to the WHO terminology, (2) associated virus, such as Epstein-Barr virus (EBV) or Kaposi sarcoma–associated virus/human herpes virus 8 (HHV8), if any, and (3) the specific immunodeficiency background (Table 1). Standardization of the nomenclature provides a nonhierarchical approach to group diagnoses in which lymphoproliferative disorders (LPDs) with similar morphologic, immunophenotypic, and genetic features from different immunodeficiency backgrounds can be classified together. This approach does not necessarily assign causality to the immunodeficiency setting or to the associated virus but recognizes the clinical context in which the LPDs arise and prompts further consideration of appropriate risk and/or alternative clinical management as necessary. For the purposes of this review, we focused our comments primarily on EBV- and HHV8-associated LPDs.
Proposed unifying nomenclature and examples of immunodeficiency-associated LPDs
| 3-Part unifying nomenclature | ||
|---|---|---|
| Name of lesion | Viral status | Specific immunodeficiency setting |
| B-cell hyperplasia (eg, plasmacytic hyperplasia) | eg, EBV+/−, HHV8+/− | eg, Posttransplant (solid organ), iatrogenic (methotrexate), immune senescence |
| Polymorphic B-cell lymphoproliferations (eg, mucocutaneous ulcer) | ||
| Lymphoma (WHO terminology) (eg, diffuse large B-cell lymphoma, Anaplastic large cell lymphoma, ALK−) | ||
| 3-Part unifying nomenclature | ||
|---|---|---|
| Name of lesion | Viral status | Specific immunodeficiency setting |
| B-cell hyperplasia (eg, plasmacytic hyperplasia) | eg, EBV+/−, HHV8+/− | eg, Posttransplant (solid organ), iatrogenic (methotrexate), immune senescence |
| Polymorphic B-cell lymphoproliferations (eg, mucocutaneous ulcer) | ||
| Lymphoma (WHO terminology) (eg, diffuse large B-cell lymphoma, Anaplastic large cell lymphoma, ALK−) | ||
| Examples of diagnostic labels | |
|---|---|
| Proposed unifying nomenclature | WHO 2016 nomenclature |
| Plasmacytic hyperplasia, EBV+, posttransplant (solid organ) | Plasmacytic hyperplasia, nondestructive posttransplant LPD |
| Polymorphic B-LPD, EBV+, iatrogenic (methotrexate) | Polymorphic LPD resembling polymorphic posttransplant LPD |
| Mucocutaneous ulcer, EBV+, primary immunodeficiency (CHARGE syndrome) | EBV+ mucocutaneous ulcer |
| Diffuse large B-cell lymphoma, EBV−, HIV infection | Diffuse large B-cell lymphoma |
| Diffuse large B-cell lymphoma, T-cell/histiocyte-rich, EBV+, immune senescence | EBV+ diffuse large B-cell lymphoma |
| Primary effusion lymphoma, HHV8+, EBV+, HIV infection | Primary effusion lymphoma |
| Examples of diagnostic labels | |
|---|---|
| Proposed unifying nomenclature | WHO 2016 nomenclature |
| Plasmacytic hyperplasia, EBV+, posttransplant (solid organ) | Plasmacytic hyperplasia, nondestructive posttransplant LPD |
| Polymorphic B-LPD, EBV+, iatrogenic (methotrexate) | Polymorphic LPD resembling polymorphic posttransplant LPD |
| Mucocutaneous ulcer, EBV+, primary immunodeficiency (CHARGE syndrome) | EBV+ mucocutaneous ulcer |
| Diffuse large B-cell lymphoma, EBV−, HIV infection | Diffuse large B-cell lymphoma |
| Diffuse large B-cell lymphoma, T-cell/histiocyte-rich, EBV+, immune senescence | EBV+ diffuse large B-cell lymphoma |
| Primary effusion lymphoma, HHV8+, EBV+, HIV infection | Primary effusion lymphoma |
Shared features of IA-LPDs in different settings and their clinical consequences
The morphologic range of LPDs arising in the background of immunodeficiency is best described in the posttransplant setting. PTLDs are typically EBV-associated B-cell proliferations, although T- and NK-cell proliferations, as well as EBV− IA-LPDs, are also recognized. As a prototype, EBV-associated B-cell IA-LPDs will be discussed in further detail because they exemplify a similar morphologic range in different immunodeficiency settings (Figure 1). This range includes B-cell hyperplasias, polymorphic B-LPDs, indolent B-cell lymphomas, aggressive B-cell lymphomas, and classic Hodgkin lymphoma–like proliferations. Therefore, a unified nomenclature is feasible, although the clinical consequences and specific treatment options for IA-LPDs may differ according to the specific immunodeficiency setting. Among the various factors contributing to the pathogenesis of IA-LPDs, some may be shared, whereas others may be specific to the immunodeficiency setting (eg, genotoxicity of previous multiagent chemotherapy in iatrogenic immunodeficiency). A common framework and vocabulary will allow these shared and setting-specific determinants of IA-LPDs to be further studied and better understood.
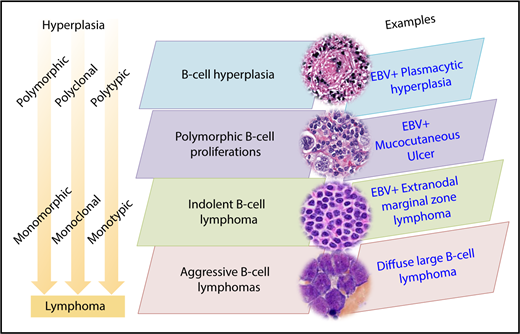
EBV-associated LPDs disorders in immunodeficiency settings
As proposed in the unifying nomenclature, the diagnostic criteria for EBV-associated IA-LPDs are applicable to all immunodeficiency settings and are summarized in Table 2. Similarities and differences among different immunodeficiency settings and their clinical implications are discussed below.

Immunodeficiency-associated B-cell hyperplasias
Immunodeficiency-associated B-cell hyperplasias have a nonspecific morphologic appearance that overlaps with other reactive conditions. Therefore, these nondestructive lesions require the presence of EBV for their association with immunodeficiency to be recognized. A relationship with immunodeficiency is debatable when EBV is negative, because no marker other than EBV indicates an association with immunodeficiency. EBV+ B-cell hyperplasias have been reported in PTLDs and iatrogenic LPDs and may also be observed in settings in which immunodeficiency is less obvious, such as in elderly patients.8 Hyperplasias that occur in the HIV setting are discussed later (HHV8-associated LPDs). Even when small B-cell clones or simple karyotypic abnormalities are present,9 most cases of immunodeficiency-associated B-cell hyperplasias regress spontaneously or with reduction of immunosuppression (in cases in which reduction of immunosuppression is feasible). Surgical excision is often sufficient for obstructive tonsillar masses. Because hyperplasias are only rarely associated with subsequent development of more aggressive IA-LPDs, watchful management is often sufficient. Clinical correlation remains essential to avoid under- and overdiagnosis.
Immunodeficiency-associated polymorphic B-cell LPDs
In contrast to B-cell hyperplasias in the immunodeficiency setting, polymorphic B-LPDs are destructive lesions that exhibit effacement of tissue architecture. In most instances they contain monoclonal B-cell receptor gene rearrangements. The morphology spans all stages of B-cell development, with a variable mixture of large B cells and Hodgkin-like cells. This range of morphologic features distinguishes polymorphic IA-B-LPDs from non-Hodgkin B-cell lymphomas (termed “monomorphic” in the WHO classification). This range of morphologic features should also prompt consideration of an underlying immunodeficiency if it is not already known. The clinical approach to polymorphic B-LPDs may vary according to the specific immunodeficiency setting. Most cases of polymorphic IA-B-LPDs in the solid organ transplant and iatrogenic clinical settings respond to reduction or withdrawal of immunosuppression, whereas the lesions in HIV+ individuals may respond to the initiation of highly active antiretroviral therapy. However, when the transplanted organ cannot be put at risk for rejection or the underlying cause of immunosuppression cannot be corrected (such as primary immunodeficiency and immune senescence), select nonresponsive or high-risk patients may require more aggressive treatment, such as immunotherapy, chemotherapy, or radiation therapy.
The recently recognized EBV+ mucocutaneous ulcer (MCU) is worthy of mention because it shares several features in common with polymorphic IA-B-LPDs.10 These are well-circumscribed often painful ulcerating lesions in mucosal or cutaneous sites that do not form a mass. EBV+ MCUs are composed of a polymorphous cell population often admixed with large B cells or Hodgkin-like cells, as seen in polymorphic PTLDs. Up to 50% of cases exhibit monoclonal B-cell receptor gene rearrangements.10 They can mimic Hodgkin or non-Hodgkin lymphomas histologically; however, their clinical presentation in mucosal sites or the skin should aid in making the correct diagnosis. Most EBV+ MCUs regress spontaneously or with reduction of immunosuppression where the latter option is possible. Rare cases may exhibit a relapsing and remitting course without further progression. In the posttransplant setting, the use of immunotherapy (rituximab) has proved effective for the clinical management of EBV+ MCUs.11
Immunodeficiency-associated indolent B-cell lymphomas
Small B-cell lymphomas in immunodeficiency settings are likely frequently coincidental rather than causally related.1 The presence of EBV is necessary for their association with immunodeficiency to be recognized. Immunodeficiency-associated small B-cell lymphomas are EBV+ plasmacytoid/plasmacytic proliferations, such as extranodal marginal zone lymphoma, lymphoplasmacytic lymphoma, and extraosseous plasmacytoma.12 The morphologic overlap with polymorphic B-LPDs is extensive and may represent a true biological continuum.4 It is possible that some small B-cell lymphomas in the immunodeficiency setting would be optimally managed with treatment strategies used for polymorphic B-LPDs.
Immunodeficiency-associated aggressive B-cell lymphomas
Aggressive B-cell lymphomas in immunodeficiency settings are usually, although not always, EBV+ and can generally be named according to their closest counterparts in immunocompetent patients. These include entities classified as monomorphic PTLDs in the current WHO classification, including diffuse large B-cell lymphoma (DLBCL), high-grade B-cell lymphoma, not otherwise specified (NOS), Burkitt lymphoma, plasmablastic lymphoma, and classic Hodgkin lymphoma (CHL). EBV− IA-B-LPDs also occur but can only be convincingly recognized in obvious immunodeficiency settings and are estimated to account for 20% to 40% of PTLDs.13 Although aggressive B-LPDs can be recognized because of their distinctive pathologic features, their association with an underlying immunodeficiency may be missed when EBV is absent. Differences in EBV+ and EBV− large B-cell lymphomas in the posttransplant setting have also been reported,14 although additional large-scale studies will be needed to validate these findings.
Similar aggressive B-cell lymphomas occur not just in the posttransplant setting but across all immunodeficiency settings. Some characteristics vary by immunodeficiency setting: for example, EBV+ Burkitt lymphoma in the setting of HIV typically has a plasmacytoid appearance. Nevertheless, the diagnostic features and aggressive clinical behavior are otherwise similar. Currently, in the posttransplant setting, these lymphomas would be grouped under monomorphic PTLDs and then subclassified accordingly. Our proposed nomenclature recognizes similar entities across the spectrum of IA-LPDs.
IA-B-LPDs are diseases in which the traditional distinction between polymorphic and monomorphic LPDs is ambiguous and nonreproducible.4 In immunodeficiency-associated large B-cell lymphomas in which there is a mixed background with increased T cells and histiocytes, classification as Hodgkin lymphoma vs non-Hodgkin lymphoma is often difficult and ambiguous. These lymphomas represent a histopathologic and immunophenotypic continuum between T-cell/histiocyte-rich large B-cell lymphoma, CHL, and EBV+ MCUs. It is clinically important to recognize the spectrum of proliferations with features of CHL in immunodeficiency settings, because they are likely to impact the choice of treatment strategies.
HHV8-associated LPDs
HHV8+ lesions have well-defined nomenclature and disease definitions and include multicentric Castleman disease (MCD); HHV8+ DLBCL, NOS; germinotropic LPDs; primary effusion lymphoma (PEL); and extracavitary PEL (Table 3).15 These typically occur in the setting of immunodeficiency, particularly HIV infection. The characteristic LPDs associated with HHV8 are described below, but it should be appreciated that there are unusual cases within the spectrum of HHV8 and HHV8/EBV LPDs with overlapping features that do not fulfill criteria for the established entities.16,17 In situations in which an HHV8-related LPD is a consideration, immunostaining for HHV8-associated latent protein LANA1 (ORF73) readily highlights the nuclei of the infected cells.
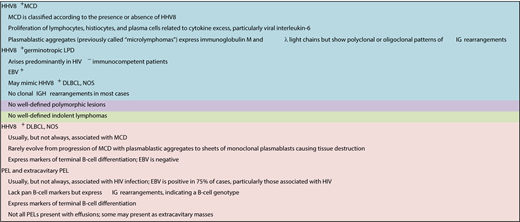
Polyclonal HHV8+ B-cell lesions
Similar to the EBV-associated B-cell hyperplasias in immunodeficiency settings, a parallel can be drawn with HHV8+ lesions, such as HHV8+ MCDs and germinotropic LPDs. These typically occur in the setting of immunodeficiency, particularly HIV infection, with the exception of germinotropic LPDs, of which most cases are HIV−. They are usually polyclonal lymphoid proliferations that show minimal, if any, destruction of the underlying architecture, similar to the EBV+ nondestructive hyperplastic lesions described in the posttransplant setting.18,19 Treatment approaches are largely driven by the clinical setting, with some cases of HHV8 MCD requiring clinical intervention, such as antiretroviral therapy, rituximab, and/or interleukin-6/interleukin-6 receptor–blocking antibodies.20
HHV8+ lymphomas
These lesions include HHV8+ DLBCLs, NOS, which are solely HHV8+, and PELs/extracavitary PELs, which are usually, but not always, HHV8+ and EBV+.21 HHV8+ DLBCL, NOS is very rare, usually arises in, and may represent progression of HHV8 MCD.15,18,19 PELs/extracavitary PELs are usually EBV+ and HHV8+ and may also arise in patients with a history of MCD.22 Although these lesions preferentially arise in HIV+ individuals, they can arise in other immunodeficiency settings, such as in elderly individuals from endemic regions and in transplant recipients.23 Aggressive lymphomas with the highest association with EBV and HHV8 tend to exhibit plasmacytic differentiation.24,25 Therefore, although there is a wider spectrum of virally associated aggressive IA-LPDs, there is still a relative degree of restriction to lesions that are derived from terminally differentiated B cells.
T-cell and NK-cell lymphomas associated with immunodeficiency
EBV is a major factor in most IA-LPDs of B-cell derivation; however, T- and NK-cell lymphomas are less frequently seen in the setting of immunodeficiency and are less often associated with EBV. Although late-occurring T-cell lymphomas in the posttransplant setting have been described,13,26-35 IA-T-LPDs do not readily fit into an orderly framework as described for IA-B-LPDs. Thus, this proposed paradigm may provide a pathway for further study of these lesions.
Only a few specific examples of associations of immunodeficiency and T-cell lymphomas are known. In recent years, an increased risk for hepatosplenic T-cell lymphoma, usually of γ-δ subtype, has been recognized, primarily in young patients with Crohn’s disease receiving immunosuppressive therapy with thiopurines and anti–tumor necrosis factor-α agents, mainly infliximab.36,37 Notably, a lesser risk is seen with other immune disorders, such as rheumatoid arthritis, treated with similar compounds. The data suggest that the risk for hepatosplenic T-cell lymphoma may be related to the synergistic effects of chronic immune stimulation and immunosuppression related to Crohn’s disease. Specific associations of T-cell lymphoproliferative disease with primary immune deficiencies are seen but are also rare. The best-documented association is a risk for T-cell leukemia, primarily T-cell prolymphocytic leukemia, in the setting of ataxia telangiectasia.38 There is a significant lack of data regarding T- and NK-cell proliferations, and progress in the field will require additional investigations across all immunodeficiency settings.
When to suspect underlying immunodeficiency
The importance of recognizing underlying immunodeficiency is clear when one considers the difference in clinical strategy between systemic chemotherapy and a graded approach, beginning with reduction of immunosuppression where possible. Communication is paramount, between the patient and the clinician to elicit the corresponding history, as well as between the clinician and the pathologist to ensure proper ancillary testing and interpretation. Relevant history includes type of transplant (solid organ, allogeneic, autologous hematopoietic stem cell transplant) and type of immunosuppressive regimen; rheumatologic and autoimmune disease; careful drug history, including prior chemotherapy; personal or family history suspicious for a primary immunodeficiency; and personal history of LPDs, which may provide a clue to underlying immunodeficiency.
Patients with rheumatologic or autoimmune disease may have increased risk for EBV+ lymphoproliferations due to the combined effect of the rheumatologic/autoimmune disease and immunosuppressive agents, such as methotrexate. In addition to well-known immunosuppressive medications, newer agents may have novel pleiotropic immune modulatory effects. For example, dasatinib, a tyrosine kinase inhibitor used in the treatment of chronic myeloid leukemia and other malignancies, is sometimes associated with clonal NK- and T-cell large granular lymphocyte proliferations,39 as well as lymphadenopathy associated with characteristic atypical reactive follicular hyperplasia.40 With increasing numbers of novel agents entering clinical practice, care must be taken not to overdiagnose malignancy in the face of lymphoproliferations that may be benign and self-resolving. Long-term effects of other profound insults to the immune system, such as history of chemotherapy, are still poorly understood, but they may also predispose patients to potentially self-resolving EBV-associated B-cell lymphoproliferations similar to those in other iatrogenic immunodeficiency settings.41,42
Manifestations of primary immunodeficiency vary markedly and are beyond the scope of this perspective; in addition to susceptibility to infection, there may be immune dysregulation, including autoimmune phenomena, and predilection to hemophagocytic lymphocytosis.43 Among patients with common variable immunodeficiency, those with autoimmune cytopenias are at increased risk for LPDs.44 The full range of EBV+ B-cell lymphoproliferations seen in the posttransplant setting have been reported in patients with primary immunodeficiency.7 Benign expansions of T-cell subsets, such as cytotoxic T cells in common variable immunodeficiency and double-negative T cells in autoimmune lymphoproliferative syndrome, are a manifestation of the underlying immunodeficiency and must not be mistaken for overt T-cell lymphoma.45,46
Practical aspects regarding the diagnosis of IA-LPDs
Given the clinical and histopathologic heterogeneity of IA-LPDs, a single biopsy, particularly a needle core, may not be representative. Excisional biopsy is preferred for diagnosis, and for mucosal or cutaneous lesions, a deep biopsy is recommended so that the characteristic architectural features of EBV+ MCUs can be appreciated. Flow immunophenotyping, if performed, may be helpful in identifying a clonal proliferation. The need for fresh tissue for flow immunophenotyping should be communicated to the surgeon so that the specimen can be divided between formalin and a suitable media, such as RPMI 1640. Molecular clonality studies may be performed on formalin-fixed paraffin-embedded tissue if indicated; however, the presence of clonal IGH or TCR gene rearrangements must be interpreted with caution in IA-LPDs because they are not synonymous with malignancy. However, investigations for genetic alterations are helpful in better characterizing these lesions.
A pathologic diagnosis suggestive of immunodeficiency provides a second opportunity to identify a potentially immunodeficiency-associated process when this is not immediately evident from the provided clinical history. In those instances, clinicians should be alerted to perform serum viral load studies by EBV DNA polymerase chain reaction to find specific support for EBV reactivation. As is evident from the discussion above, certain lesions, such as EBV+ MCUs or EBV+ polymorphic B-LPDs, are usually an indication of defective immune surveillance for EBV and likely underlying immunodeficiency of varied etiology.
A comprehensive biological framework for IA-LPDs
The genesis of IA-LPDs is multifactorial and may include chronic antigenic stimulation, overproduction of cytokines, altered immune checkpoints, and increased propensity to DNA damage. At least in some clinical scenarios, there is evidence that shared pathogenetic mechanisms underlie IA-LPDs. In many instances, EBV and HHV8 are important drivers, irrespective of the immunodeficiency setting. The significance of immunosuppression to lymphomagenesis is even less well understood in cases in which the virus is lacking. HIV is known to contribute to lymphomagenesis due to its immunosuppressive effect, but a direct role in lymphomagenesis has also been described.47-49
Recent investigations show that 9p24.1 copy number alterations and upregulation of PD-L1 are common in EBV+ and EBV− IA-LPDs arising in diverse immunodeficiency states, including PTLDs, HIV, iatrogenic immunodeficiency, and immune senescence.4,50-53 Interestingly, this finding suggests a possible common and specific role for immune checkpoint blockade as an effective treatment strategy for IA-LPDs. These findings underscore the rationale for an overarching framework that includes all IA-LPDs.
It should be appreciated that the nature of the underlying immunodeficiency and the clinical setting will impact the character of the IA-LPD and its clinical management. Moreover, some IA-LPDs are relatively unique to certain clinical settings. For example, mycophenolate mofetil has been shown to specifically predispose to the development of primary central nervous system DLBCL, in which calcineurin exerts a protective effect.54 Awareness of these specific associations is necessary to elicit the appropriate history and to guide clinical management.
Conclusions
In the 50 years since the first description of PTLDs,55 recognition of IA-LPDs has expanded to include a spectrum of lesions occurring in diverse immunodeficiency settings. Some categories of lesions and some immunodeficiency settings are better understood than others, and newer lesions and contexts continue to emerge. However, this expanded spectrum of IA-LPDs has come at the expense of nonuniform cumbersome terminology and inconsistently applied disease definitions that have hampered progress in the field. In this perspective, we have outlined a conceptual framework to unify and standardize nomenclature and disease definitions across categories of underlying immunodeficiency. Lymphoma classifications have previously used the strategy of adopting unifying terminology such that problematic lesions that span the boundary between ≥2 distinct entities can be better studied. Highly heterogeneous groups have benefitted from such an approach, which has provided improved clarity to bring back to the clinic. The unifying nomenclature, by providing a common framework, is expected to guide future iterations of terminology, disease definitions, and classification for all IA-LPDs. This approach, in turn, may guide the choice of optimal therapies for patients while still recognizing the specific clinical context and treatment requirements of each immunodeficiency setting.
Acknowledgments
The authors thank their hematology and oncology colleagues Sven DeVos (University of California Los Angeles), Wyndham Wilson (National Cancer Institute), Ranjana Advani (Stanford University School of Medicine), Joseph Sparano (Albert Einstein College of Medicine), and Pam McKay (University of Glasgow) for their excellent feedback and discussion regarding the clinical usefulness of our proposal.
Authorship
Contribution: All authors actively participated in writing this article, including writing parts of the original text, contributing to discussions, editing drafts, and providing input for tables and figures.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Daphne de Jong, VU University Medical Center, Department of Pathology, De Boelelaan 1117, 1081HV Amsterdam, The Netherlands; e-mail: daphne.dejong@vumc.nl; and Yasodha Natkunam, Stanford University, School of Medicine, 300 Pasteur Dr, Stanford, CA 94305; e-mail: yaso@stanford.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal