

Key Points
Overexpression of GATA6 induces aberrant CD137L expression on tumor cells of CTCL.
CD137-CD137L interactions promote cell proliferation and migration in CTCL cells, representing potential therapeutic targets.

Abstract
CD137 and its ligand, CD137L, are expressed on activated T cells and antigen-presenting cells, respectively. Recent studies have shown that CD137L and CD137 are aberrantly expressed by tumor cells, especially in some hematopoietic malignancies, and interactions between these molecules on tumor cells promote tumor growth. In this study, we investigated the roles of CD137L and CD137 in cutaneous T-cell lymphoma (CTCL), represented by mycosis fungoides and Sézary syndrome. Flow cytometric analysis showed that primary Sézary cells and CTCL cell lines (Hut78, MyLa, HH, SeAx, and MJ) aberrantly expressed CD137L. CD137L expression by tumor cells in CTCL was also confirmed by immunohistochemistry. Anti-CD137L–neutralizing antibody inhibited proliferation, survival, CXCR4-mediated migration, and in vivo growth in CTCL cell lines through inhibition of phosphorylation of AKT, extracellular signal-regulated kinase 1/2, p38 MAPK, and JNK. Moreover, suppression of CD137L signaling decreased antiapoptotic proteins Bcl-2 and phosphorylated Bad. We also explored the transcription factor regulating CD137L expression. Because GATA6 has been proposed as an oncogene in many types of tumors with aberrant CD137L expression, we examined GATA6 expression and the involvement of GATA6 in CD137L expression in CTCL. DNA hypomethylation and histone acetylation induced GATA6 overexpression in CTCL cells. Furthermore, chromatin immunoprecipitation, luciferase reporter assay, and knockdown by short hairpin RNA showed that GATA6 directly upregulated CD137L expression. Inhibition of GATA6 resulted in decreased survival and in vivo growth in CTCL cells. Collectively, our findings prompt a novel therapeutic approach to CTCL based on the discovery that the GATA6/CD137L axis plays an important role in the tumorigenesis of CTCL.
Introduction
Mycosis fungoides (MF) and Sézary syndrome (SS), the most common types of cutaneous T-cell lymphoma (CTCL),1 are characterized by proliferation of mature CD4+ T-helper cells.2 MF typically presents in the form of skin patches and/or plaques, which can progress to skin tumors, with subsequent involvement of lymph nodes, peripheral blood, and visceral organs. In some cases, skin lesions become confluent and finally develop into erythroderma without blood involvement. SS is defined by the triad of generalized erythroderma, lymphadenopathy, and circulating atypical T cells.1 Patients with advanced stages of MF and SS have poor prognosis; currently, there is no standard treatment of these patients.3,4 Like other lymphomas, cell-to-cell interactions between tumor cells contribute to survival and regulate malignant growth in the form of autocrine or paracrine loops in CTCL. In addition to various cytokines, such as interleukin-13 (IL-13), IL-15, and IL-32,5-7 surface proteins including CD40 and CD28 provide the direct molecular bridge between tumor cells and adjacent cells, resulting in tumor progression.8-10
CD137 is a transmembrane glycoprotein of the tumor necrosis factor receptor superfamily and is broadly expressed on immune cell populations, including activated T cells and natural killer (NK) cells.11 CD137 has diverse roles in the immune response and its most arrestive function is to enhance the proliferation, survival, and cytokine production of T cells and NK cells, leading to enhancement of antitumor effects.11-14 Indeed, agonistic anti-CD137 monoclonal antibody has been shown to be highly effective in eliminating tumors via activating CD8+ T cells and NK cells.11,14-17 The ligand of CD137, CD137L, is mainly expressed by antigen-presenting cells.18 In addition to its ability to costimulate T cells by triggering CD137 receptor, CD137L signals back into antigen-presenting cells themselves, resulting in increased adherence, cytokine production, activation marker expression, and survival.18,19 In some carcinomas, aberrant CD137L expression is detected on tumor cells, suggesting that CD137L may influence the progression of tumors.20,21 Moreover, in some hematopoietic malignancies, such as Hodgkin lymphoma and leukemias, both CD137 and CD137L are expressed on malignant cells and CD137-CD137L interactions promote tumor growth.22-24 These results suggest that CD137-CD137L interactions play an important role not only in antitumor immunity but also in tumor progression, indicating that blockade of CD137-CD137L interactions can be exploited for therapy in some malignancies. Thus, it is important to know the involvement of CD137-CD137L interactions in each malignancy before clinical application of agonistic anti-CD137 antibody or blockade of CD137-CD137L interactions. Concerning CTCL, although it is reported that CD137 is expressed on tumor cells in most MF cases,25 so far, CD137L expression has not been studied.
In this report, we demonstrated that both CD137L and CD137 were expressed on CTCL cells and that CD137L messenger RNA (mRNA) expression levels in CTCL lesional skin were associated with increased disease-specific mortality. Moreover, suppression of CD137-CD137L interactions by neutralizing antibodies inhibited cell proliferation, survival, migration, and in vivo growth. Blocking CD137L decreased phosphorylation of AKT, extracellular signal-regulated kinase 1/2 (ERK1/2), p38 MAPK, and JNK as well as antiapoptotic proteins (Bcl-2 and phosphorylated Bad [p-Bad]). Furthermore, we also showed that GATA6 directly regulated aberrant CD137L expression. Thus, the GATA6/CD137L axis may have a critical role in the tumorigenesis of CTCL.
Materials and methods
Patients and samples
As summarized in supplemental Table 1 (available on the Blood Web site), skin samples were collected from 88 patients with CTCL (63 male and 25 female patients; mean plus or minus standard deviation [SD] age: 58.2 ± 14.5 years; 20 cases of patch MF, 19 cases of plaque MF, 29 cases of tumor MF, 9 cases of erythrodermic MF, and 11 cases of SS) and 20 healthy controls (51.9 ± 15.1 years). Blood samples were collected from 43 patients with CTCL (28 male and 15 female patients; mean plus or minus SD age: 58.0 ± 16.7 years; 11 cases of patch MF, 9 cases of plaque MF, 12 cases of tumor MF, 4 cases of erythrodermic MF, and 7 cases of SS) and 15 healthy controls (51.8 ± 14.9 years). As previously described,26 peripheral blood mononuclear cells (PBMCs) were obtained from 10 SS patients and 14 healthy controls (mean plus or minus SD age: 63.9 ± 13.6 and 51.4 ± 18.5 years, respectively; supplemental Table 2). All patients with MF and SS were given diagnoses according to International Society for Cutaneous Lymphomas (ISCL)/European Organization of Research and Treatment of Cancer (EORTC) criteria.1,27 All samples were collected after informed consent during daily clinical practice. The medical ethical committee of the University of Tokyo approved all described studies and the study was conducted according to the Declaration of Helsinki principles.
Analysis of CD137L, CD137, CXCR4, and GATA6 expression in lesional skin and sera of CTCL or CTCL cells
Quantitative reverse transcription polymerase chain reaction (RT-PCR) and immunohistochemistry for CD137L, CD137, and GATA6 were performed using lesional skin of CTCL. Specific enzyme-linked immunosorbent assays for soluble CD137L (sCD137L) and sCD137 were performed using sera of CTCL patients. Flow cytometric analysis for CD137, CD137L, and CXCR4 were performed using CTCL cells including CTCL cell lines and CD4+CD7− malignant T cells from SS patients. Western blotting for GATA6 was performed using CTCL cells. Detailed methods are given in supplemental Methods.
Analysis of the effects of CD137-CD137L interactions on CTCL cells
After CTCL cells were treated with the anti-CD137L– or anti-CD137–neutralizing antibody, proliferation assays, apoptosis assays, in vivo animal experiments, intracellular flow cytometric analysis, western blotting, and a migration assay were performed. Gene knockdown of CD137L was also performed. Detailed methods are given in supplemental Methods.
Analysis of CD137L gene regulation by GATA6
A chromatin immunoprecipitation (ChIP) assay for GATA6 occupancy of CD137L promoters and gene knockdown of GATA6 were performed using CTCL cells. A luciferase reporter assay using CD137L promoter constructs and expression vector for GATA6 was performed using HEK293 cells. Detailed methods are given in supplemental Methods.
Analysis of epigenetic regulation of GATA6 gene in CTCL cells
After CTCL cells were treated with 5-Aza-2′-deoxycytidine (5-aza; Cayman Chemical) or FK228 (Gloucester Pharmaceuticals), GATA6 expression was examined. A ChIP assay for acetylated histone occupancy of GATA6 promoters and a methylation-specific PCR (MSP) assay were performed using CTCL cells. Detailed methods are given in supplemental Methods.
Statistical analysis
The GraphPad Prism 7.01 software program was used for statistical analyses. All in vitro experiments were repeated at least 3 times and mean plus or minus SD was determined. Statistical analysis between 2 groups was performed using the Welch t test. For comparing 2 group values that did not follow Gaussian distribution, a 2-tailed Mann-Whitney U test was used. A paired t test was used to determine significant differences between anti-CD137L–neutralizing antibody-treated and control groups when using patient tumor cell samples. Correlation coefficients were determined by using the Spearman rank correlation test. P values of < .05 were considered statistically significant.
Results
CD137L expression on malignant T cells in CTCL
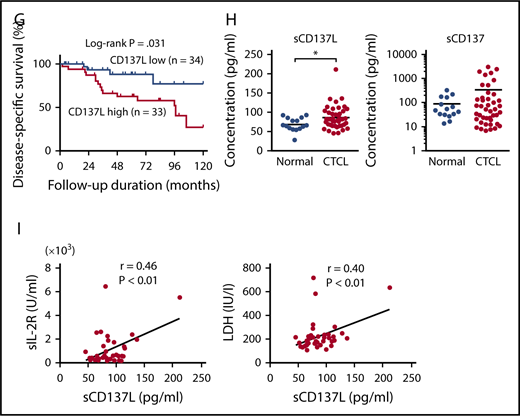
To assess CD137L and CD137 expression on tumor cells of CTCL, we first examined CD137L and CD137 expression on PBMCs from SS patients and healthy controls by flow cytometry. CD4+CD7− malignant T cells from SS patients expressed both CD137L and CD137, whereas most of CD4+CD7+ and CD8+ T cells expressed only CD137 (Figure 1A-B). By contrast, CD4+ or CD8+ T cells from healthy controls barely expressed CD137L (Figure 1A-B). In SS patients, the percentage of CD137L+ and CD137+ cells correlated with each other (Figure 1B). We also found that human CTCL cell lines (Hut78, MyLa, HH, SeAx, and MJ cells) and a murine T-cell lymphoma cell line (EL-4) expressed both CD137L and CD137 (Figure 1C). Because CD137 is reported to be expressed on tumor cells of MF by immunohistochemistry similar to our results,25 we next immunolabeled samples of CTCL lesional skin for CD137L. As shown in Figure 1D, CD137L was expressed on a part of tumor cells. Moreover, CD137L and CD137 mRNA expression levels in CTCL lesional skin were significantly higher than those in healthy skin (Figure 1E), consistent with the results of flow cytometry and immunohistochemistry. In addition, CD137L and CD137 mRNA levels significantly correlated with each other in CTCL lesional skin (r = 0.61, P < .0001; Figure 1F). We next examined whether CD137L mRNA levels correlated with patient survival using Kaplan-Meier analysis and the log-rank test. A strong correlation was found between high CD137L mRNA levels (>0.0014) and increased disease-related death (P = .031; log-rank test hazard ratio, 2.97; Figure 1G), although these patients were not treated uniformly but in a variety of ways according to their stage and conditions. Thus, CTCL cells expressed both CD137L and CD137, and CD137L mRNA levels in lesional skin correlated with disease-specific mortality, suggesting that aberrant CD137L expression is associated with development of CTCL.
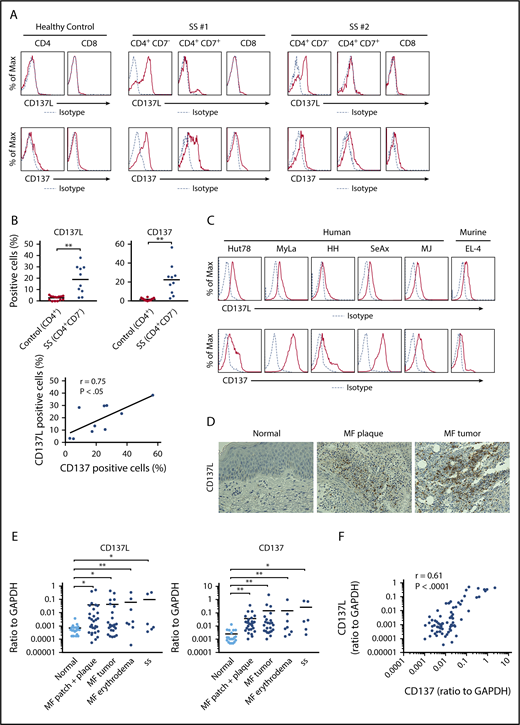
CD137L expression on malignant T cells in CTCL. (A-C) CD137L and CD137 expression was analyzed by flow cytometry in CD4+CD7−, CD4+CD7+, and CD8+ T cells from 10 SS patients, CD4+ and CD8+ T cells from 14 healthy controls (A-B), human CTCL cell lines (Hut78, MyLa, SeAx, HH, and MJ cells), and murine lymphoma cell line EL-4 cells (C). Representative flow cytometric plots are shown (A, C). The percentage of CD137L+ or CD137+ cells was counted in CD4+CD7− T cells from SS patients or CD4+ T cells from healthy controls. Correlations between the percentage of CD137L+ and CD137+ cells in CD4+CD7− T cells were examined (B). (D) CD137L expression in lesional skin of CTCL was determined by immunohistochemistry (original magnification ×400). Diaminobenzidine was used for visualizing the staining and counterstaining with Mayer hematoxylin was performed. Representative results are shown. (E) Quantitative RT-PCR was performed to measure expression levels of CD137L and CD137 using mRNA extracted from CTCL lesional skin (n = 57; 51 MF cases and 6 SS cases) and healthy skin (n = 20). (F) Correlations between CD137L and CD137 mRNA levels in CTCL lesional skin. (G) Kaplan-Meier analysis for disease-specific survival of 57 CTCL patients with high CD137L mRNA levels (>0.0014) and those with low CD137L mRNA levels (≤0.0014). (H) Serum sCD137L and sCD137 levels in CTCL patients (n = 43). (I) Correlations between serum sCD137L levels and sIL-2 receptor (sIL-2R) or lactate dehydrogenase levels (LDH) levels in CTCL patients. Means are presented as bars. *P < .05, **P < .01. GAPDH, glyceraldehyde-3-phosphate dehydrogenase; Max, maximum.
CD137L expression on malignant T cells in CTCL. (A-C) CD137L and CD137 expression was analyzed by flow cytometry in CD4+CD7−, CD4+CD7+, and CD8+ T cells from 10 SS patients, CD4+ and CD8+ T cells from 14 healthy controls (A-B), human CTCL cell lines (Hut78, MyLa, SeAx, HH, and MJ cells), and murine lymphoma cell line EL-4 cells (C). Representative flow cytometric plots are shown (A, C). The percentage of CD137L+ or CD137+ cells was counted in CD4+CD7− T cells from SS patients or CD4+ T cells from healthy controls. Correlations between the percentage of CD137L+ and CD137+ cells in CD4+CD7− T cells were examined (B). (D) CD137L expression in lesional skin of CTCL was determined by immunohistochemistry (original magnification ×400). Diaminobenzidine was used for visualizing the staining and counterstaining with Mayer hematoxylin was performed. Representative results are shown. (E) Quantitative RT-PCR was performed to measure expression levels of CD137L and CD137 using mRNA extracted from CTCL lesional skin (n = 57; 51 MF cases and 6 SS cases) and healthy skin (n = 20). (F) Correlations between CD137L and CD137 mRNA levels in CTCL lesional skin. (G) Kaplan-Meier analysis for disease-specific survival of 57 CTCL patients with high CD137L mRNA levels (>0.0014) and those with low CD137L mRNA levels (≤0.0014). (H) Serum sCD137L and sCD137 levels in CTCL patients (n = 43). (I) Correlations between serum sCD137L levels and sIL-2 receptor (sIL-2R) or lactate dehydrogenase levels (LDH) levels in CTCL patients. Means are presented as bars. *P < .05, **P < .01. GAPDH, glyceraldehyde-3-phosphate dehydrogenase; Max, maximum.
Serum sCD137L levels in patients with CTCL
To evaluate the involvement of soluble forms of CD137L and CD137 in development of CTCL, we measured serum sCD137L and sCD137 levels in patients with CTCL. Serum sCD137L levels were significantly increased in patients with CTCL, whereas serum sCD137 levels in CTCL patients were comparable to those in healthy controls (Figure 1H). Additionally, serum sCD137L levels positively correlated with serum levels of sIL-2 receptor and lactate dehydrogenase in CTCL patients (Figure 1I). Considering that sCD137L can bind to CD137 expressed on healthy T cells, leading to release of inflammatory cytokines,28,29 sCD137L in sera of CTCL patients may play roles in development of CTCL. The modest increase of sCD137L in sera of CTCL patients and the great overlap between healthy controls and CTCL patients (Figure 1H), however, suggested that CD137-CD137L interactions mainly mediate their survival benefit through cell-cell interactions among tumor cells.
The blockade of CD137-CD137L interactions inhibits proliferation, survival, and in vivo growth of CTCL cells
To assess the function of CD137-CD137L interactions on CTCL cells, we blocked CD137L and CD137 using neutralizing antibodies. Either anti-CD137L or anti-CD137 antibody significantly decreased the proliferation of CTCL cell lines and the combination of both antibodies had a similar effect to single administration of either antibody (Figure 2A; supplemental Figure 1A). The frequency of cell cluster formation was not changed after anti-CD137L antibody administration. We further examined effects of murine anti-CD137L and anti-CD137 antibodies on EL-4 cells, and they suppressed the proliferation of EL-4 cells (Figure 2B). We also found that anti-CD137L antibody inhibited proliferation of CD4+CD7− malignant T cells from SS patients; by contrast, the effect was not observed in CD4+ T cells from healthy controls or CD4+CD7+ T cells from SS patients (Figure 2C). In addition, anti-CD137L antibody induced apoptosis in CTCL cell lines and EL-4 cells (Figure 2D-E; supplemental Figure 1B). We next stimulated the cells using plate-coated recombinant CD137L protein according to previous reports.23,29 Recombinant CD137L protein promoted proliferation of CTCL cells (Figure 2F). To assess the in vivo effects of anti-CD137L antibody, we used a xenograft model.30 Treatment with anti-CD137L antibody significantly decreased tumor formation by Hut78 and HH cells in NOD/SCID interleukin-2 receptor γ-chain-deficient (NSG) mice or SCID-Beige mice in vivo (Figure 2G; supplemental Figure 1C). Because the immune system is severely impaired in those mice, we also determined effects of anti-CD137L antibody on EL-4 cells using C57BL/6 mice, whose T cells and B cells were functionally normal. Significantly smaller tumors were formed in the mice treated with anti-CD137L antibody, compared with those treated with phosphate-buffered saline (PBS; Figure 2H). Thus, blocking of CD137-CD137L interactions suppressed CTCL cell proliferation and survival in vitro and tumor growth in vivo.
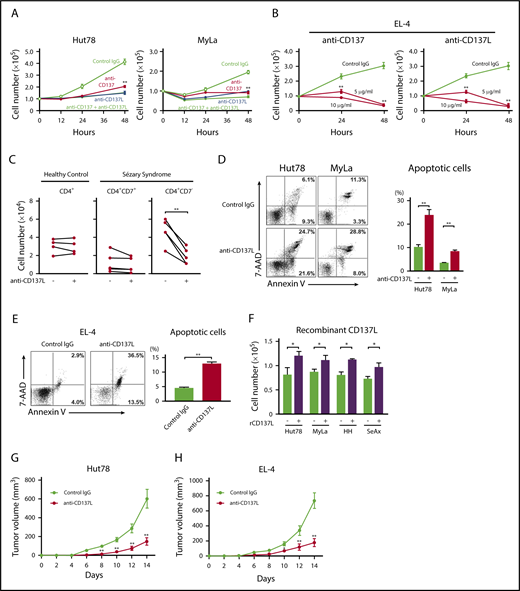
The blockade of CD137-CD137L interactions inhibits proliferation, survival, and in vivo growth of CTCL tumor cells. (A-B) Hut78 and MyLa cells (1.0 × 105 per well) were cultured with anti-CD137 (5 μg/mL) and/or anti-CD137L (5 μg/mL) neutralizing antibodies for 48 hours (A). EL-4 cells (1.0 × 105 per well) were cultured with anti-CD137 antibody (5 or 10 μg/mL) or anti-CD137L–neutralizing antibody (5 or 10 μg/mL) for 48 hours (B). Viable cells were counted at indicated time points by trypan blue exclusion. Statistical difference was compared with isotype-treated cells. (C) PBMCs from 5 SS patients and 4 age-matched healthy controls (1.0 × 105 per well) were cultured with anti-CD137L–neutralizing antibody (5 μg/mL) for 48 hours, and the percentage of CD4+CD7− and CD4+CD7+ T cells in SS patients or CD4+ T cells from healthy controls were determined by flow cytometry. Viable cells were counted at 48-hour time points by trypan blue exclusion. Statistical difference was compared with isotype-treated cells (paired Student t test). (D-E) Hut78, MyLa, and EL-4 cells (1.0 × 105 per well) were cultured with anti-CD137L–neutralizing antibody (5 μg/mL) for 24 hours. Apoptosis (Annexin V+ and 7-AAD−) was evaluated with Annexin V and 7-aminoactinomycin D (7-AAD) staining for flow cytometric analysis. (F) Hut78, MyLa, HH, and SeAx cells (5.0 × 104 per well) were cultured with plate-coated recombinant CD137L (10 μg/mL) for 48 hours. Viable cells were counted. Data are presented as mean plus or minus SD. *P < .05, **P < .01 (A-F). (G-H) Hut78 cells (5.0 × 106) were injected into NSG mice with anti-CD137L–neutralizing antibody (50 μg/mL) or isotype immunoglobulin G (IgG) (G). EL-4 cells (3.0 × 106) were injected into C57BL/6 mice with anti-CD137L–neutralizing antibody (50 μg/mL) or isotype IgG (H). Each reagent was injected on days 4, 7, and 11. The tumor volume was calculated using the equation: V = π (L1 × L22)/6, where V = volume (mm3), L1 = longest diameter (mm), and L2 = shortest diameter (mm). Data are presented as means plus standard error of the mean (SEM; n = 12). *P < .05, ** P < .01 by Mann-Whitney U test compared with control group.
The blockade of CD137-CD137L interactions inhibits proliferation, survival, and in vivo growth of CTCL tumor cells. (A-B) Hut78 and MyLa cells (1.0 × 105 per well) were cultured with anti-CD137 (5 μg/mL) and/or anti-CD137L (5 μg/mL) neutralizing antibodies for 48 hours (A). EL-4 cells (1.0 × 105 per well) were cultured with anti-CD137 antibody (5 or 10 μg/mL) or anti-CD137L–neutralizing antibody (5 or 10 μg/mL) for 48 hours (B). Viable cells were counted at indicated time points by trypan blue exclusion. Statistical difference was compared with isotype-treated cells. (C) PBMCs from 5 SS patients and 4 age-matched healthy controls (1.0 × 105 per well) were cultured with anti-CD137L–neutralizing antibody (5 μg/mL) for 48 hours, and the percentage of CD4+CD7− and CD4+CD7+ T cells in SS patients or CD4+ T cells from healthy controls were determined by flow cytometry. Viable cells were counted at 48-hour time points by trypan blue exclusion. Statistical difference was compared with isotype-treated cells (paired Student t test). (D-E) Hut78, MyLa, and EL-4 cells (1.0 × 105 per well) were cultured with anti-CD137L–neutralizing antibody (5 μg/mL) for 24 hours. Apoptosis (Annexin V+ and 7-AAD−) was evaluated with Annexin V and 7-aminoactinomycin D (7-AAD) staining for flow cytometric analysis. (F) Hut78, MyLa, HH, and SeAx cells (5.0 × 104 per well) were cultured with plate-coated recombinant CD137L (10 μg/mL) for 48 hours. Viable cells were counted. Data are presented as mean plus or minus SD. *P < .05, **P < .01 (A-F). (G-H) Hut78 cells (5.0 × 106) were injected into NSG mice with anti-CD137L–neutralizing antibody (50 μg/mL) or isotype immunoglobulin G (IgG) (G). EL-4 cells (3.0 × 106) were injected into C57BL/6 mice with anti-CD137L–neutralizing antibody (50 μg/mL) or isotype IgG (H). Each reagent was injected on days 4, 7, and 11. The tumor volume was calculated using the equation: V = π (L1 × L22)/6, where V = volume (mm3), L1 = longest diameter (mm), and L2 = shortest diameter (mm). Data are presented as means plus standard error of the mean (SEM; n = 12). *P < .05, ** P < .01 by Mann-Whitney U test compared with control group.
The blockade of CD137-CD137L interactions impairs CXCR4-mediated migration of CTCL cells
CD137L mediates upregulation of CXCR4 expression in dendritic cells.31 In addition, it is well known that CXCR4 is expressed on CTCL cells and is associated with skin homing of tumor cells.32-34 Thus, we examined whether CD137-CD137L interactions induced CXCR4 on CTCL cells, using anti-CD137L–neutralizing antibody. Anti-CD137L antibody significantly downregulated CXCR4 mRNA and surface protein expression in Hut78 and Myla cells (Figure 3A-B). In addition, as shown in Figure 3C, treatment with anti-CD137L antibody significantly inhibited CXCL12/CXCR4-mediated migration. Interestingly, anti-CD137L antibody and AMD3100, a CXCR4 antagonist, did not have a synergic effect on CXCL12/CXCR4-mediated migration of Hut78 cells (supplemental Figure 2). We also showed that suppression of CD137L inhibited CXCR4 surface expression in CD4+CD7− malignant T cells from SS patients (n = 6; Figure 3D). Thus, blocking of CD137-CD137L interactions downregulated CXCR4 expression and CXCL12-induced migration of CTCL cells.
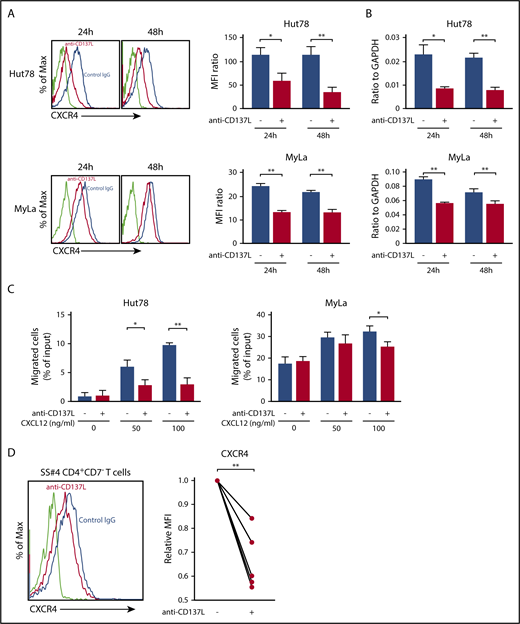
The blockade of CD137-CD137L interactions impairs CXCR4-mediated migration of CTCL tumor cells. (A-B) Hut78 and MyLa cells (1.0 × 106 per well) were cultured with anti-CD137L–neutralizing antibody (5 μg/mL) for 24 or 48 hours. CXCR4 expression was evaluated by flow cytometry (A), and quantitative RT-PCR (B). The mean fluorescent intensity (MFI) ratio was determined as MFI of target molecule/MFI of isotype control. (C) Hut78 and MyLa cells were treated with anti-CD137L–neutralizing antibody (5 μg/mL) for 12 hours, and then assessed in a migration assay for 12 hours at 37°C with human CXCL12 protein (0, 50, or 100 ng/mL). The percentage of migrating cells relative to input was determined. (D) PBMCs from 6 SS patients (1.0 × 106 per well) were cultured with anti-CD137L antibody (5 μg/mL) for 24 hours. CXCR4 expression on CD4+CD7− T cells was evaluated by flow cytometry. MFI expression was normalized to the MFI expression of isotype-treated cells. Representative results are shown. Data are presented as mean plus or minus SD. *P < .05, **P < .01
The blockade of CD137-CD137L interactions impairs CXCR4-mediated migration of CTCL tumor cells. (A-B) Hut78 and MyLa cells (1.0 × 106 per well) were cultured with anti-CD137L–neutralizing antibody (5 μg/mL) for 24 or 48 hours. CXCR4 expression was evaluated by flow cytometry (A), and quantitative RT-PCR (B). The mean fluorescent intensity (MFI) ratio was determined as MFI of target molecule/MFI of isotype control. (C) Hut78 and MyLa cells were treated with anti-CD137L–neutralizing antibody (5 μg/mL) for 12 hours, and then assessed in a migration assay for 12 hours at 37°C with human CXCL12 protein (0, 50, or 100 ng/mL). The percentage of migrating cells relative to input was determined. (D) PBMCs from 6 SS patients (1.0 × 106 per well) were cultured with anti-CD137L antibody (5 μg/mL) for 24 hours. CXCR4 expression on CD4+CD7− T cells was evaluated by flow cytometry. MFI expression was normalized to the MFI expression of isotype-treated cells. Representative results are shown. Data are presented as mean plus or minus SD. *P < .05, **P < .01
The blockade of CD137-CD137L interactions suppresses phosphorylation of AKT, ERK1/2, p38 MAPK, and JNK, and expression of Bcl-2 and p-Bad
CD137-CD137L signaling is known to induce phosphorylation of AKT, ERK1/2, p38 MAPK, and JNK in various types of cells.35-37 Consistent with those studies, western blot analysis showed that anti-CD137L antibody decreased phosphorylation of AKT, ERK1/2, p38 MAPK, and JNK in Hut78 and MyLa cells, which was also confirmed by intracellular flow cytometry (Figure 4A-B). In addition, anti-CD137L antibody inhibited anti-CD3 antibody-mediated phosphorylation of AKT and ERK1/2 (supplemental Figure 3). Because anti-CD137L antibody induced apoptosis in CTCL cell lines, we next investigated the expression of antiapoptotic proteins in Hut78 and MyLa cells. Anti-CD137L antibody decreased Bcl-2 and p-Bad, both of which were reported to be upregulated via phosphorylation of AKT, ERK1/2, p38 MAPK, or JNK (Figure 4A-B).38-43 We also analyzed effects of CD137L suppression in CD4+CD7− malignant T cells from 4 SS patients by intracellular flow cytometry. After treatment of anti-CD137L antibody, expression levels of p-AKT, p-ERK1/2, p-p38 MAPK, p-JNK, Bcl-2, and p-Bad were significantly inhibited in CD4+CD7− malignant T cells from SS patients, but not in CD4+ T cells from healthy controls (Figure 4C). Consistent with these results showing that multiple pathways are initiated by CD137-CD137L interactions, anti-CD137L–neutralizing antibody and Bcl-2 inhibitor (ABT-737) additively inhibited proliferation and survival of Hut78 cells (supplemental Figure 4), suggesting that CD137-CD137L interactions induce proliferation of CTCL cells through not only Bcl-2 pathways but also other pathways. Collectively, anti-CD137L–neutralizing antibody suppressed phosphorylation of multiple signaling pathways (AKT, ERK1/2, p38 MAPK, and JNK) and antiapoptotic proteins (Bcl-2 and p-Bad) in CTCL cells.
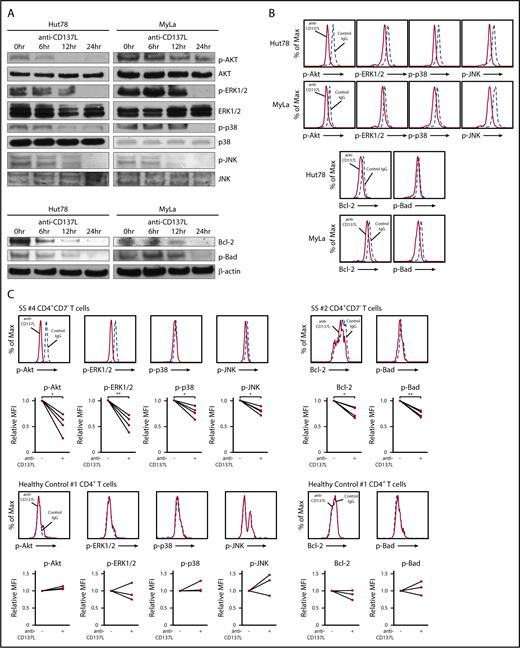
The blockade of CD137-CD137L interactions suppresses phosphorylation of AKT, ERK1/2, p38 MAPK, and JNK, and expression of Bcl-2 and p-Bad. (A) Western blotting analysis was conducted on the lysates of Hut78 and MyLa cells treated with anti-CD137L–neutralizing antibody (5 μg/mL) or isotype control for 0, 6, 12, or 24 hours. Phosphorylation of AKT, ERK1/2, p38 MAPK, and JNK, and expression of Bcl-2 and p-Bad were measured. (B-C) Hut78, MyLa cells, and PBMCs from 4 SS patients as well as 3 healthy controls were treated with anti-CD137L–neutralizing antibody (5 μg/mL) or isotype control for 24 hours. Expression levels of p-AKT, p-ERK1/2, p-p38 MAPK, p-JNK, Bcl-2, and p-Bad were determined by intracellular flow cytometry in Hut78, MyLa cells (B), CD4+CD7− T cells from PBMCs of SS patients and CD4+ T cells from healthy controls (C). The MFI expression was normalized against the MFI expression of isotype-treated cells. Representative results are shown. *P < .05, **P < .01
The blockade of CD137-CD137L interactions suppresses phosphorylation of AKT, ERK1/2, p38 MAPK, and JNK, and expression of Bcl-2 and p-Bad. (A) Western blotting analysis was conducted on the lysates of Hut78 and MyLa cells treated with anti-CD137L–neutralizing antibody (5 μg/mL) or isotype control for 0, 6, 12, or 24 hours. Phosphorylation of AKT, ERK1/2, p38 MAPK, and JNK, and expression of Bcl-2 and p-Bad were measured. (B-C) Hut78, MyLa cells, and PBMCs from 4 SS patients as well as 3 healthy controls were treated with anti-CD137L–neutralizing antibody (5 μg/mL) or isotype control for 24 hours. Expression levels of p-AKT, p-ERK1/2, p-p38 MAPK, p-JNK, Bcl-2, and p-Bad were determined by intracellular flow cytometry in Hut78, MyLa cells (B), CD4+CD7− T cells from PBMCs of SS patients and CD4+ T cells from healthy controls (C). The MFI expression was normalized against the MFI expression of isotype-treated cells. Representative results are shown. *P < .05, **P < .01
The knockdown of CD137L attenuates cell proliferation, survival, and CXCR4 expression
To confirm that CD137-CD137L interactions promote cell growth, survival, and CXCR4 expression of CTCL cells, Hut78 cells were transduced with CRISPR-Cas9 single-guided RNA (sgRNA) against CD137L (sgCD137L) or control scrambled sgRNA (sgSCR). Knockdown of CD137L by 2 sgRNA was confirmed by flow cytometric analysis and quantitative RT-PCR (Figure 5A). Stable suppression of CD137L resulted in significantly reduced cell growth and survival compared with control cells (Figure 5B-C). We also showed that phosphorylation of AKT, ERK1/2, p38 MAPK, and JNK was decreased in Hut78-sgCD137L cells (Figure 5D). Moreover, significantly smaller tumors were formed by Hut78-sgCD137L cells compared with those by Hut78-sgSCR cells, when the cells were injected into NSG mice (Figure 5E). In addition, flow cytometric analysis and quantitative RT-PCR showed that surface protein and mRNA expression levels of CXCR4 were decreased in CD137L-knockdown Hut78 cells (Figure 5F). Thus, knockdown of CD137L expression attenuated cell proliferation, survival, and CXCR4 expression via downregulation of p-AKT, p-ERK1/2, p-p38 MAPK, and p-JNK.
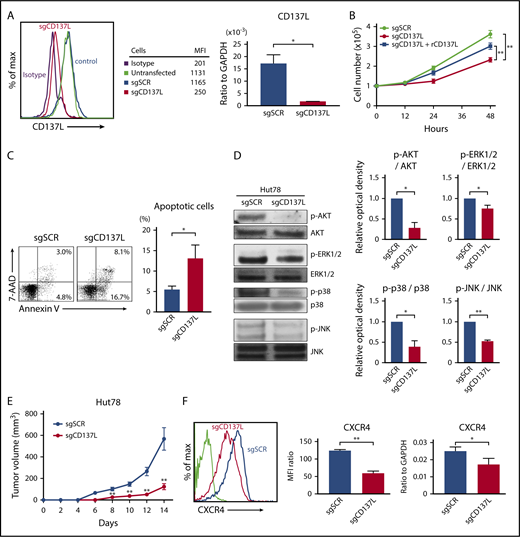
The knockdown of CD137L attenuates cell proliferation, survival, and CXCR4 expression. (A-E) Hut78 cells were transfected with either CD137L-targeting sgRNA (sgCD137L) or control scrambled sgRNA (sgSCR). (A) CD137L expression levels were assayed by flow cytometry and quantitative RT-PCR. MFIs are shown for a representative experiment. (B) Hut78-sgCD137L and Hut78-sgSCR cells (1.0 × 105 per well) were cultured with or without plate-coated recombinant CD137L (10 μg/mL) for 48 hours. Viable cells were counted at indicated time points. (C) Apoptosis (Annexin V+ and 7-AAD−) was evaluated with Annexin V and 7-AAD staining for flow cytometric analysis in Hut78-sgCD137L and Hut78-sgSCR cells. (D) Western blotting analysis was conducted on the lysates of Hut78-sgCD137L and Hut78-sgSCR cells. Phosphorylation of AKT, ERK1/2, p38 MAPK, and JNK was measured. The representative blot from 3 independent experiments. Band intensities were calculated by Image J software. Relative optical density of p-AKT/AKT, p-ERK1/2/ERK1/2, p-p38 MAPK/p38 MAPK, and p-JNK/JNK was calculated and normalized to the value of Hut78-sgSCR cells. Data are presented as mean plus or minus SD. *P < .05, **P < .01. (E) Hut78-sgCD137L or Hut78-sgSCR cells (5.0 × 106) were injected into NSG mice. The tumor volume was calculated using the equation: V = π (L1 × L22)/6, where V = volume (mm3), L1 = longest diameter (mm), and L2 = shortest diameter (mm). Values are means plus or minus SEM (n = 12). **P < .01 by Mann-Whitney U test compared with control group. (F) CXCR4 expression was evaluated by flow cytometry, and quantitative RT-PCR. Representative results are shown. MFI expression was normalized to the MFI expression of isotype-treated cells. Data are presented as mean plus or minus SD. *P < .05, **P < .01.
The knockdown of CD137L attenuates cell proliferation, survival, and CXCR4 expression. (A-E) Hut78 cells were transfected with either CD137L-targeting sgRNA (sgCD137L) or control scrambled sgRNA (sgSCR). (A) CD137L expression levels were assayed by flow cytometry and quantitative RT-PCR. MFIs are shown for a representative experiment. (B) Hut78-sgCD137L and Hut78-sgSCR cells (1.0 × 105 per well) were cultured with or without plate-coated recombinant CD137L (10 μg/mL) for 48 hours. Viable cells were counted at indicated time points. (C) Apoptosis (Annexin V+ and 7-AAD−) was evaluated with Annexin V and 7-AAD staining for flow cytometric analysis in Hut78-sgCD137L and Hut78-sgSCR cells. (D) Western blotting analysis was conducted on the lysates of Hut78-sgCD137L and Hut78-sgSCR cells. Phosphorylation of AKT, ERK1/2, p38 MAPK, and JNK was measured. The representative blot from 3 independent experiments. Band intensities were calculated by Image J software. Relative optical density of p-AKT/AKT, p-ERK1/2/ERK1/2, p-p38 MAPK/p38 MAPK, and p-JNK/JNK was calculated and normalized to the value of Hut78-sgSCR cells. Data are presented as mean plus or minus SD. *P < .05, **P < .01. (E) Hut78-sgCD137L or Hut78-sgSCR cells (5.0 × 106) were injected into NSG mice. The tumor volume was calculated using the equation: V = π (L1 × L22)/6, where V = volume (mm3), L1 = longest diameter (mm), and L2 = shortest diameter (mm). Values are means plus or minus SEM (n = 12). **P < .01 by Mann-Whitney U test compared with control group. (F) CXCR4 expression was evaluated by flow cytometry, and quantitative RT-PCR. Representative results are shown. MFI expression was normalized to the MFI expression of isotype-treated cells. Data are presented as mean plus or minus SD. *P < .05, **P < .01.
GATA6 acts as an oncogenic gene in CTCL cells through directly regulating CD137L expression
To identify the possible transcription factor inducing aberrant CD137L expression on CTCL cells, we analyzed the promoter region of the human CD137L gene (gene ID: 8744) using the MOTIF searching programs (Tfsitescan, http://www.ifti.org/Tfsitescan/; JASPAR, http://jaspar.genereg.net/). Among transcription factors that can bind to the promoter region of CD137L, we focused on GATA6 because GATA6 mRNA levels are reported to be elevated in malignant T cells in SS patients compared with healthy controls.44 Similar to that study, we found that tumor cells in CTCL patients abundantly expressed GATA6, whereas infiltrating lymphocytes in healthy controls were weakly positive for GATA6 by immunohistochemistry (Figure 6A). The number of GATA6+ cells in all stage of CTCL skin was significantly higher than that of healthy skin (Figure 6B). In addition, GATA6 mRNA levels in CTCL lesional skin were higher than those in healthy skin (Figure 6C). Importantly, GATA6 mRNA levels positively correlated with those of CD137L (r = 0.66, P < .0001; Figure 6D). Moreover, western blotting analysis revealed that GATA6 protein levels were highly increased in PBMCs from SS patients and CTCL cell lines, compared with PBMCs from healthy controls (Figure 6E). Notably, in CTCL cell lines, GATA6 and CD137L mRNA levels were strongly correlated with each other (Figure 6F). Such positive correlations between GATA6 and CD137L expression prompted us to investigate whether GATA6 directly regulated CD137L expression in CTCL cells. ChIP analysis displayed the binding of GATA6 to the CD137L promoter at −345/−152 bp upstream of the transcription start site in Hut78 and MyLa cells (Figure 6G). Furthermore, the luciferase reporter assay using HEK293 cells revealed that GATA6 overexpression increased CD137L reporter activity in a dose-dependent manner (Figure 6H). To determine whether GATA6 positively regulates CD137L expression, we suppressed GATA6 expression in Hut78 and MyLa cells by short hairpin RNA (shRNA). Knockdown of GATA6 was confirmed by western blotting analysis and quantitative RT-PCR (Figure 6I-J). Flow cytometric analysis and RT-PCR showed that stable suppression of GATA6 resulted in significantly reduced CD137L expression levels, followed by downregulation of CXCR4 (Figure 6K-L). As expected, we also found that GATA6 suppression led to decreased phosphorylation of AKT and ERK1/2 as well as cell growth, and induced apoptosis (Figure 6M-O). Furthermore, NSG mice injected with Hut78 cells transfected with GATA6 shRNA formed significantly smaller tumors compared with those transfected with scrambled shRNA (Figure 6P). Thus, GATA6, whose expression was elevated in CTCL cells, directly induced aberrant CD137L expression.
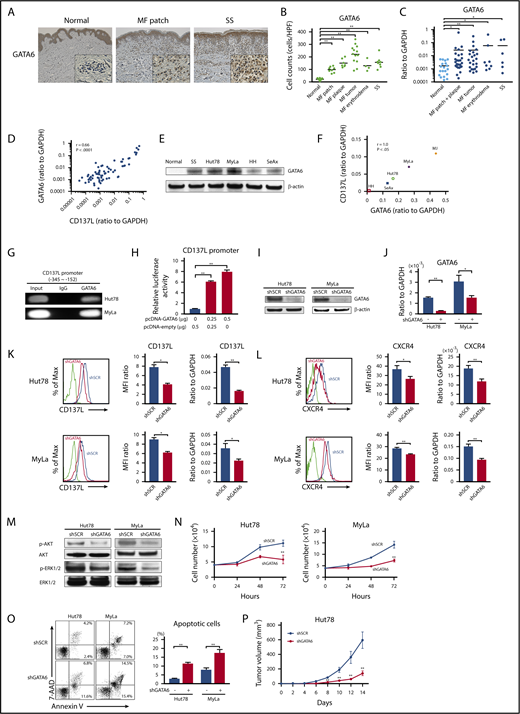
GATA6 acts as an oncogenic gene in CTCL tumor cells through directly regulating CD137L expression. (A) GATA6 expression in lesional skin of CTCL was determined by immunohistochemistry. Diaminobenzidine was used for visualizing the staining and counterstaining with Mayer hematoxylin was performed. Representative results are shown (original magnification ×100). Insets, High-magnification images (original magnification ×400). (B) GATA6+ cells were counted per high-power field (×400). (C) Quantitative RT-PCR was performed to measure expression levels of GATA6 using mRNA extracted from CTCL lesional skin (n = 57; 51 MF cases and 6 SS cases) and healthy skin (n = 20). (D) A correlation between GATA6 and CD137L mRNA levels in CTCL lesional skin. (E) Western blotting analysis for GATA6 protein expression was conducted on the lysates of PBMCs from 1 SS patient as well as healthy controls and CTCL cell lines. (F) A correlation between GATA6 and CD137L mRNA levels in CTCL cell lines. (G) ChIP assay in Hut78 and MyLa cells was performed with anti-GATA6 antibody and the primer specific for the designed area of the CD137L gene promoter. (H) Luciferase reporter assay using CD137L promoter constructs. HEK293 cells were cotransfected with CD137L promoter constructs and GATA6 expression vector. Forty-eight hours after transfection, luciferase activity was measured. (I-P) Hut78 and MyLa cells were transfected with either GATA6-targeting shRNA (shGATA6) or control scrambled shRNA (shSCR). (I-J) GATA6 expression levels were assayed by western blotting analysis (I) and quantitative RT-PCR (J). (K-L) CD137L (K) and CXCR4 (L) expression levels were determined by flow cytometric analysis and quantitative RT-PCR. Representative results are shown. (M) Western blotting analysis was conducted on the lysates of shGATA6 and shSCR cells. Expression levels of p-AKT and p-ERK1/2 were measured. (N) shGATA6 or shSCR transduced cells (4.0 × 104 per well) were cultured for 72 hours. Viable cells were counted at indicated time points by trypan blue exclusion. (O) Apoptosis (Annexin V+ and 7-AAD−) in shGATA6 and shSCR cells was evaluated with Annexin V and 7-AAD staining for flow cytometric analysis. Data are presented as mean plus or minus SD. *P < .05, **P < .01. (P) Hut78-shGATA6 or Hut78-shSCR cells (5.0 × 106) were injected into NSG mice. The tumor volume was calculated using the equation: V = π (L1 × L22)/6, where V = volume (mm3), L1 = longest diameter (mm), and L2 = shortest diameter (mm). Values are means plus or minus SEM (n = 12). *P < .05, **P < .01 by the Mann-Whitney U test compared with control group.
GATA6 acts as an oncogenic gene in CTCL tumor cells through directly regulating CD137L expression. (A) GATA6 expression in lesional skin of CTCL was determined by immunohistochemistry. Diaminobenzidine was used for visualizing the staining and counterstaining with Mayer hematoxylin was performed. Representative results are shown (original magnification ×100). Insets, High-magnification images (original magnification ×400). (B) GATA6+ cells were counted per high-power field (×400). (C) Quantitative RT-PCR was performed to measure expression levels of GATA6 using mRNA extracted from CTCL lesional skin (n = 57; 51 MF cases and 6 SS cases) and healthy skin (n = 20). (D) A correlation between GATA6 and CD137L mRNA levels in CTCL lesional skin. (E) Western blotting analysis for GATA6 protein expression was conducted on the lysates of PBMCs from 1 SS patient as well as healthy controls and CTCL cell lines. (F) A correlation between GATA6 and CD137L mRNA levels in CTCL cell lines. (G) ChIP assay in Hut78 and MyLa cells was performed with anti-GATA6 antibody and the primer specific for the designed area of the CD137L gene promoter. (H) Luciferase reporter assay using CD137L promoter constructs. HEK293 cells were cotransfected with CD137L promoter constructs and GATA6 expression vector. Forty-eight hours after transfection, luciferase activity was measured. (I-P) Hut78 and MyLa cells were transfected with either GATA6-targeting shRNA (shGATA6) or control scrambled shRNA (shSCR). (I-J) GATA6 expression levels were assayed by western blotting analysis (I) and quantitative RT-PCR (J). (K-L) CD137L (K) and CXCR4 (L) expression levels were determined by flow cytometric analysis and quantitative RT-PCR. Representative results are shown. (M) Western blotting analysis was conducted on the lysates of shGATA6 and shSCR cells. Expression levels of p-AKT and p-ERK1/2 were measured. (N) shGATA6 or shSCR transduced cells (4.0 × 104 per well) were cultured for 72 hours. Viable cells were counted at indicated time points by trypan blue exclusion. (O) Apoptosis (Annexin V+ and 7-AAD−) in shGATA6 and shSCR cells was evaluated with Annexin V and 7-AAD staining for flow cytometric analysis. Data are presented as mean plus or minus SD. *P < .05, **P < .01. (P) Hut78-shGATA6 or Hut78-shSCR cells (5.0 × 106) were injected into NSG mice. The tumor volume was calculated using the equation: V = π (L1 × L22)/6, where V = volume (mm3), L1 = longest diameter (mm), and L2 = shortest diameter (mm). Values are means plus or minus SEM (n = 12). *P < .05, **P < .01 by the Mann-Whitney U test compared with control group.
DNA hypomethylation and histone acetylation contribute to GATA6 overexpression in CTCL cells
Generally speaking, DNA methylation represses gene expression and histone acetylation enhances gene expression.45,46 It is reported that promoter-specific hypomethylation is associated with overexpression of GATA6 in SS tumor cells.44 We further investigated whether GATA6 expression was epigenetically promoted in tumor cells of CTCL including MF. CTCL cell lines were treated with 2 epigenetic inhibitors, 5-aza (a DNA methyltransferase inhibitor) and FK228 (a histone deacetylase inhibitor), leading to an increase in GATA6 expression (supplemental Figure 5A). As for DNA methylation, MSP revealed that certain CpG islands in the GATA6 promoter were unmethylated in CTCL cell lines, whereas they were predominantly methylated in healthy CD4+ cells (supplemental Figure 5B). Furthermore, regarding histone acetylation, ChIP assay indicated that histone H3 and H4 on the GATA6 promoter were more acetylated in CTCL cells than healthy CD4+ cells (supplemental Figure 5C). Although GATA6 is epigenetically overexpressed in CTCL cell lines and 5-aza and FK228 further increased GATA6 expression as described, both inhibitors suppressed proliferation of CTCL cells and their combination had an additive effect (supplemental Figure 6A), similar to the previous report.47 5-aza promoted CXCR4 expression and CXCR4-mediated migration, whereas FK228 suppressed both of them (supplemental Figure 6B-C), consistent with previous reports.48-50 The effect of their combination was different depending on CTCL cell lines: their combination promoted CXCR4 expression and migration in Hut78 cells and suppressed both of them in MyLa cells. These results are probably due to various genes other than GATA6 that are regulated by these drugs. Thus, GATA6 is epigenetically overexpressed in CTCL cells by DNA hypomethylation and histone deacetylation.
Discussion
In this study, we first revealed that CTCL cells aberrantly expressed CD137L in addition to CD137. Similar to our results, aberrant CD137L expression is reported in some carcinomas.20,21 CD137L expressed on hepatocellular carcinoma cell lines was regarded as functional because IL-8 production was induced after ligation with CD137.21 Moreover, acute myeloid leukemia cells stimulated the release of immunosuppressive cytokine IL-10 via CD137L signaling.51 These previous reports have suggested that CD137L expression on tumor cells contributes to tumor development in some malignancies, as IL-8 and IL-10 are associated with tumor growth or antitumor immune suppression.52-54 We also found that CD137L promoted proliferation and survival of CTCL cells through multiple signaling pathways and that CD137L mRNA expression levels in lesional skin correlated with increased risk of disease-related death. These results suggest that CD137L expression on CTCL cells is largely involved in development of CTCL like other malignancies with CD137L expression. In dendritic cells, CD137L signaling mediates migration via upregulation of CXCR4 and CCR7.31 Although CCR7 is expressed on SS tumor cells but not on MF tumor cells,55 CXCR4 has been reported to be expressed in both MF and SS tumor cells.32-34 Thus, we assessed whether aberrant CD137L expression influenced CXCR4-mediated migration in CTCL. Anti-CD137L–neutralizing antibody inhibited CXCR4 expression and migration of CTCL cells. Considering that inhibition of p38 MAPK activity reduces CXCR4 expression in breast cancer,56 our result suggests that CD137L expression on CTCL cells promotes CXCR4-mediated migration, possibly through the p38 MAPK–signaling pathway. Collectively, CD137-CD137L interactions play an advantageous role in CTCL and can be exploited for targeted therapy of the disease.
Regarding CD137-CD137L interaction-targeted therapy, agonistic anti-CD137 monoclonal antibody is thought to be a promising therapy for malignancies based on the rationale that CD137 signaling can enhance antitumor activity of T cells and NK cells.14 Indeed, clinical trials of 2 agonistic antibodies, urelumab and utomilumab, for several malignancies including melanoma, non–small lung cell cancer, and non-Hodgkin B-cell lymphoma, have been designed and implemented.14,16,17 However, such immunotherapies exhibit an almost dichotomous response pattern. Clinical responses are usually achieved in only a fraction of patients with a majority of patients showing minimal or no appreciable benefit. That is because the effect is dependent on the status of antitumor immunity in each patient. Thus, many efforts have been made to increase the fraction of patients who respond to immunotherapy in several ways, such as a combination of 2 immunotherapies, a combination with standard therapy, and identifying the patient who will respond to immunotherapies by evaluating antitumor immunity.57,58 On the other hand, different from the situation in many malignancies, our results and previous reports revealed that both CD137 and CD137L were expressed on tumor cells of some hematopoietic malignancies including CTCL, leukemias, and Hodgkin lymphoma, and that CD137-CD137L interactions promoted the proliferation of tumor cells.22-24 This effect is considered to be independent of the status of antitumor immunity. Thus, blockade of CD137-CD137L interactions might be a superior therapy to immunotherapies in malignancies that express both CD137 and CD137L, although it may attenuate antitumor immunity of CD8+ T cells and NK cells. Actually, we showed that anti-CD137L–neutralizing antibody had the therapeutic effect on not only human CTCL cell growth in immunodeficient mice but also tumor formation of mouse T-cell lymphoma in C57BL/6 mice.
Previous studies show that CD137L protein is not detected on normal T cells, or only at low levels, and that activated T cells express only CD137.59,60 On the other hand, malignant T cells of CTCL expressed both CD137 and CD137L. Our results indicate that tumor cells in CTCL acquire CD137L expression during the transformation process like other cancers. We focused on GATA6, which is a member of the GATA transcription factor family and plays critical regulatory roles in tissue development,61 because the promoter region of CD137L has the binding motif of GATA6 and GATA6 is overexpressed in tumor cells of SS.44 In addition, GATA6 expression is associated with cell survival, cell proliferation, carcinogenesis, and prognosis in a variety of malignancies, such as colon cancer,62,63 hepatocellular carcinoma,64 and breast adenocarcinoma65 ; interestingly, many such malignancies also express CD137L.20,21 We found that GATA6 was overexpressed in CTCL cells and directly regulated CD137L expression. Moreover, we showed that GATA6 was epigenetically overexpressed in CTCL cells, resulting from DNA hypomethylation and histone acetylation. Furthermore, suppression of GATA6 induced decreased cell proliferation, survival, and tumor growth in CTCL cells. Collectively, our results indicate that GATA6 may act as an oncogenic gene and thereby contribute to tumorigenesis and tumor progression in CTCL through inducing aberrant CD137L expression.
In conclusion, CD137L was aberrantly expressed in CTCL cells, in addition to CD137. Blockade of CD137-CD137L interactions between tumor cells decreased cell proliferation, survival, migration, and in vivo growth. Moreover, blocking CD137L attenuated phosphorylation of AKT, ERK1/2, p38 MAPK, JNK, and antiapoptotic proteins (Bcl-2 and p-Bad). Epigenetically overexpressed GATA6 via DNA hypomethylation and histone acetylation directly regulated CD137L expression. Thus, the GATA6/CD137L axis and CD137-CD137L interactions may play important roles in tumorigenesis of CTCL by promoting proliferation, survival, and migration of tumor cells and thus potentially be therapeutic targets for this life-threating disease (Figure 7).
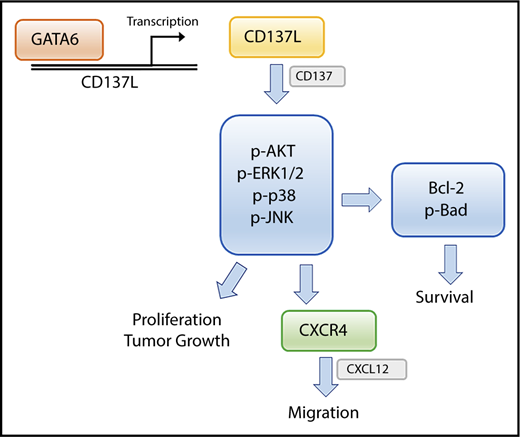
Schematic model of the main findings of this study. GATA6 directly upregulates CD137L gene transcription. CD137-CD137L interactions in CTCL cells promote cell proliferation, survival, and CXCR4-mediated migration through multiple signaling pathways.
Schematic model of the main findings of this study. GATA6 directly upregulates CD137L gene transcription. CD137-CD137L interactions in CTCL cells promote cell proliferation, survival, and CXCR4-mediated migration through multiple signaling pathways.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Kazuyasu Fujii (Department of Dermatology, Kagoshima University, Kagoshima, Japan) for providing Hut78, MyLa, HH, SeAx, and MJ cells and Sam T. Hwang (Department of Dermatology, School of Medicine, University of California Davis, Sacramento, CA) for providing EL-4 cells. The authors thank Tamami Kaga for technical assistance.
This work was supported in part by grants from the Ministry of Education, Culture, Sports, Science and Technology in Japan (16K19709).
Authorship
Contribution: H.K. designed and performed the experiments, analyzed the data, and wrote the manuscript; T.M. designed the research and experiments, analyzed the data, and wrote the manuscript; N.S.-T., R.N., T.O., and H.S. contributed to sample collection from human subjects; M.S. designed the research and analyzed the data; and S.S supervised the research and provided guidance.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Tomomitsu Miyagaki, Department of Dermatology, Graduate School of Medicine, University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8655, Japan; e-mail: asahikari1979@gmail.com.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal