

Key Points
A variable chain plasmid library enables rapid TCR cloning and expression to assess specificity and functional avidity.
Antigen-specific T-cell responses to neoantigens in CLL and after personalized neoantigen vaccine in melanoma are characterized.

Abstract
Recent studies have highlighted the promise of targeting tumor neoantigens to generate potent antitumor immune responses and provide strong motivation for improving our understanding of antigen-T-cell receptor (TCR) interactions. Advances in single-cell sequencing technologies have opened the door for detailed investigation of the TCR repertoire, providing paired information from TCRα and TCRβ, which together determine specificity. However, a need remains for efficient methods to assess the specificity of discovered TCRs. We developed a streamlined approach for matching TCR sequences with cognate antigen through on-demand cloning and expression of TCRs and screening against candidate antigens. Here, we first demonstrate the system’s capacity to identify viral-antigen-specific TCRs and compare the functional avidity of TCRs specific for a given antigen target. We then apply this system to identify neoantigen-specific TCR sequences from patients with melanoma treated with personalized neoantigen vaccines and characterize functional avidity of neoantigen-specific TCRs. Furthermore, we use a neoantigen-prediction pipeline to show that an insertion-deletion mutation in a putative chronic lymphocytic leukemia (CLL) driver gives rise to an immunogenic neoantigen mut-MGA, and use this approach to identify the mut-MGA-specific TCR sequence. This approach provides a means to identify and express TCRs, and then rapidly assess antigen specificity and functional avidity of a reconstructed TCR, which can be applied for monitoring antigen-specific T-cell responses, and potentially for guiding the design of effective T-cell-based immunotherapies.
Introduction
The clinical success of cancer immunotherapy has generated greater urgency for improving our understanding of the interaction between T-cell receptors (TCRs) and antigens. Antigen discovery has always been a high priority in the field, with a recent focus on tumor neoantigens, a class of HLA-bound peptides arising from tumor-specific mutations.1,2 On the TCR side, exciting new developments in single-cell sequencing have provided an unprecedented understanding of the clonal structure of T-cell responses.3,4 However, a streamlined approach for directly linking specific TCRs to antigens has been lacking.
The modular structure of the TCR is generated by random recombination of V, D, and J gene segments and pairing of TCRα and TCRβ chains. Early approaches to TCR repertoire analysis used techniques such as complementarity determining region 3 (CDR3) spectratyping, flow cytometry with monoclonal antibodies directed against different variable α (Vα) and β (Vβ) segments, and bulk sequencing of the TCRβ chain.5 These methods provide insight into the diversity of TCRs present in a population, overall patterns of Vα and Vβ usage, and extent of clonal expansion. More recently, single-cell sequencing technologies have enabled detailed analysis of the CDR3 region from paired TCRαβ chains, making it possible for the exquisite specificity of the TCR to be reconstructed.3,4,6 A number of methods for TCR cloning and expression have been reported,6,7 but the rich diversity of the TCR repertoire poses challenges for high-throughput reconstruction.
In this study, we leveraged the modular structure of the TCR to develop a streamlined approach to reconstruct any TCR on demand, express it on a Jurkat∆αβ reporter cell line, and match its cognate antigen from among a chosen pool of candidate antigens. We use this controlled system to assess the functional avidity of the reconstituted TCRs. Functional avidity of a T cell describes its activation threshold and is an important metric to assess for potential antitumor activity. Here, we demonstrate the pipeline’s utility to study T-cell responses specific for viral antigens and tumor neoantigens.
Methods
Human peripheral blood mononuclear cell samples and cell lines
Heparinized blood from healthy donors and study participants enrolled in a clinical trial of personalized melanoma neoantigen vaccine (NCT01970358) was obtained on Institutional Review Board-approved protocols at Dana-Farber Cancer Institute. Cell lines used included HEK 293T cells, single HLA-expressing cell lines, and a TCR-deficient Jurkat cell line (Jurkat∆αβ, genetically engineered to lack endogenous TCR expression by CRISPR-Cas9 targeting, with stable expression of CD8/CD28 or CD4/CD8/CD28).8 Jurkat∆αβ cells were transduced to express a nuclear factor of activated T cells (NFAT)-luciferase construct9,10 in PEW lentiviral vector (Jurkat∆αβ reporter cells). Luciferase expression is controlled by an inducible interleukin (IL)-2 promoter and 6 tandem repeats of NFAT binding sites (supplemental Figure 1A, available on the Blood Web site). Costimulatory molecule expression on TCR-transduced Jurkat∆αβ cells at rest and after stimulation was assessed by flow cytometry (supplemental Figure 2).
Generation, detection, and isolation of viral antigen-reactive and patient neoantigen-reactive T cells
For expansion of CEF peptide-, melanoma neoantigen-, and mut-MGA-reactive T cells, peripheral blood mononuclear cells (PBMCs) were stimulated in 24-well cell culture plates at 5 × 106 cells/well with individual or pooled peptides (each at 2 μg/mL) in the presence of IL-7 (20 ng/mL; R&D Systems). On day 3, IL-2 (20 U/mL; Amgen) was added. Half-medium change and cytokine supplementation were performed every 3 days. For mut-MGA-reactive T cells, PBMCs were additionally stimulated twice, each for 1 week, with peptides and CD4/CD8-depleted PBMCs. After 10 to 21 days, interferon (IFN)γ ELISPOT or IFNγ secretion assay were performed (supplemental Methods).
Paired TCRαβ chain single-cell sequencing
Linked TCRα/TCRβ Illumina libraries from single cells were generated using a targeted multiprimer-based approach,4 followed by Illumina sequencing (supplemental Methods).
TCR cloning and expression system
A plasmid library encoding variable chain segments was designed to enable rapid TCR cloning, such that any TCR could be assembled from 2 library components, and a double-stranded oligonucleotide encoding CDR3α and CDR3β. The variable chain plasmid library consisted of 2 types of vectors: 46 variable α with constant β (Vα-Cβ) and 52 variable β with constant α (Vβ-Cα) (supplemental Methods; supplemental Figure 1B; supplemental Table 1). For each TCR, CDR3α and CDR3β oligonucleotide flanked by BsaI restriction sites was custom synthesized (supplemental Figure 1C). Golden Gate Assembly11,12 of 2 plasmids from the library and an oligonucleotide, all digested with BsaI, produced a single vector encoding both TCRα and TCRβ, separated by a linker sequence including an F2A13 (Figure 1A). The TCR construct was assembled and transferred to lentiviral vector PEW and used to transduce Jurkat∆αβ reporter cells.

A streamlined approach for identifying and reconstructing TCRs, and testing for specificity to candidate antigens. T cells reactive against antigens are isolated by IFNγ secretion and submitted for single-cell paired TCRαβ sequencing. (A) Dominant TCR clonotypes are individually cloned by joining variable chain plasmid library components and an on-demand oligonucleotide encoding the CDR3 regions (supplemental Figure 1B-D). Linker sequence includes furin, SGSG, and F2A sequence. (B) TCRs are expressed in Jurkat∆αβ cells with an NFAT-luciferase construct (Jurkat∆αβ reporter cells; supplemental Figure 1A,E). (C) TCRs are screened for specificity against candidate antigens, with detection based on IL-2 secretion, CD69 expression, or luciferase activity.
A streamlined approach for identifying and reconstructing TCRs, and testing for specificity to candidate antigens. T cells reactive against antigens are isolated by IFNγ secretion and submitted for single-cell paired TCRαβ sequencing. (A) Dominant TCR clonotypes are individually cloned by joining variable chain plasmid library components and an on-demand oligonucleotide encoding the CDR3 regions (supplemental Figure 1B-D). Linker sequence includes furin, SGSG, and F2A sequence. (B) TCRs are expressed in Jurkat∆αβ cells with an NFAT-luciferase construct (Jurkat∆αβ reporter cells; supplemental Figure 1A,E). (C) TCRs are screened for specificity against candidate antigens, with detection based on IL-2 secretion, CD69 expression, or luciferase activity.
Detection of antigen-specific TCRs and assessment of functional avidity
Autologous antigen-presenting cells (APCs, derived from CD4/CD8-depleted PBMCs) and HLA-expressing cell lines were pulsed with candidate peptides, and TCR-expressing reporter cells were cocultured with the pulsed APCs overnight. TCR activation was measured by IL-2 enzyme-linked immunosorbent assay (ELISA), luciferase activity, or CD69 expression (supplemental Methods). To measure functional avidity of antigen-specific TCRs, TCR-expressing reporter cells were cocultured with APCs pulsed with a range of peptide concentrations14 (10 pg/mL-10 μg/mL), and IL-2 was measured.
Prediction of neoantigens arising from indel mutations
Predicted neoantigens were identified in a cohort of 157 patients with CLL, from whom leukemic and matched germline DNA had previously been analyzed by whole-exome sequencing.15,16 Indels were identified using Indelocator and Strelka and reviewed by visualizing in Integrative Genomics Viewer.17 Indel validation was performed by Sanger sequencing and/or RNA sequencing. HLA typing from patients with CLL was inferred by POLYSOLVER.18 NetMHCpan v2.4, an artificial neural network-based algorithm, was used to predict the binding affinity of potential epitopes arising from these indels to HLA class I.19-21
Results
Establishing an approach for matching TCR sequences with cognate antigen
We sought to develop an approach to test the antigen specificity of TCRs identified by single-cell sequencing of paired TCRα and TCRβ chains. This method consists of 3 general steps to link antigen with specific TCR: on-demand cloning of discovered TCRs using our variable chain plasmid library, expression of TCRs in a reporter cell line that allows detection of T-cell activation, and screening of TCR-expressing reporter cells against chosen candidate antigens (Figure 1).
Critical to enabling rapid on-demand cloning, we developed a variable chain plasmid library based on the modular structure of the TCR. The library consists of 2 types of vectors (Vα-Cβ and Vβ-Cα), each encoding 1 Vα or Vβ segment with its respective leader sequence, and the opposite constant region. In total, we generated a library of 46 Vα-Cβ and 52 Vβ-Cα plasmids to encompass the diversity of the TCR repertoire (supplemental Table 1). For each discovered TCRαβ pair, double-stranded oligonucleotides approximately 280 bp in length were designed and custom synthesized to encode the CDR3 regions from TCRα and TCRβ (supplemental Table 2). BsaI restriction sites were included in the plasmids and the CDR3 oligonucleotides to facilitate accurate joining by Golden Gate Assembly. As shown in Figure 1A, 2 plasmids from the library and an oligonucleotide were assembled to generate a single plasmid vector encoding both TCRα and TCRβ, separated by an F2A to ensure balanced expression of the 2 chains. To express the reconstructed TCRs, we transferred individual plasmids into a lentiviral vector, and transduced the NFAT-luciferase TCR-deficient Jurkat cell line (Jurkat∆αβ reporter cells; Figure 1B; supplemental Figure 1A). In this way, rapid detection of antigen specificity could be achieved by measuring luminescence, surface expression of CD69, or IL-2 secretion after exposure to candidate antigens (Figure 1C).
To establish proof of concept of this system, we cloned and expressed a TCR sequence known to be specific for the HLA-A*24:02-restricted Epstein Barr virus (EBV)-EBNA3A peptide in reporter cells6 (supplemental Methods; supplemental Figure 1B-E). We verified the expression of this TCR through staining with HLA-A*24:02/EBNA3A tetramer and anti-Vβ5-1 antibody, each showing 86.9% and 97.1% positivity, respectively. In addition, although endogenous CD3 is not expressed on surface of parental Jurkat∆αβ, it is stabilized when TCR is expressed. Indeed, we observed that TCR expression resulted in 90% staining of cells with the anti-CD3 antibody (Figure 2A). To evaluate functional antigen specificity of the EBNA3A-TCR, reporter cells were cocultured with APCs (B721.221 cells expressing HLA-A*24:02) pulsed with EBNA3A peptide, or the irrelevant HLA-A*24:02-restricted BRLF1 peptide as control (supplemental Table 3A). IL-2 was produced selectively in the presence of EBNA3A peptide (Figure 2B). These results demonstrated the feasibility of using this plasmid library to readily assemble a complete TCR based on sequence information from the CDR3α/β regions.

The EBNA3A-specific TCR can be matched to its cognate peptide. (A) The EBNA3A-specific TCR (supplemental Figure 1B-E) was cloned from the plasmid library, and expressed in Jurkat∆αβ reporter cells by lentiviral transduction. Expression of the TCR on the reporter cells was verified by staining with EBNA3A tetramer, anti-Vβ5-1, and anti-CD3 antibodies. (B) Specificity of the EBNA3A-specific TCR for cognate antigen was confirmed by detection of IL-2 secretion selectively after coculture of reporter cells with B721.221-A*24:02 cells presenting EBNA3A peptide compared with control HLA-A*24:02-restricted peptide BRLF1 (P = .014; 2-sample t-test with Welch’s correction from 2 technical replicates). Phorbol 12-myristate 13-acetate (PMA)/ionomycin stimulation was used as a positive control.
The EBNA3A-specific TCR can be matched to its cognate peptide. (A) The EBNA3A-specific TCR (supplemental Figure 1B-E) was cloned from the plasmid library, and expressed in Jurkat∆αβ reporter cells by lentiviral transduction. Expression of the TCR on the reporter cells was verified by staining with EBNA3A tetramer, anti-Vβ5-1, and anti-CD3 antibodies. (B) Specificity of the EBNA3A-specific TCR for cognate antigen was confirmed by detection of IL-2 secretion selectively after coculture of reporter cells with B721.221-A*24:02 cells presenting EBNA3A peptide compared with control HLA-A*24:02-restricted peptide BRLF1 (P = .014; 2-sample t-test with Welch’s correction from 2 technical replicates). Phorbol 12-myristate 13-acetate (PMA)/ionomycin stimulation was used as a positive control.
Deconvolution of antigen targets for viral-reactive TCRs
We next sought to demonstrate that our approach could be used to identify the cognate antigen of reconstructed TCRs from among a pool of candidate antigens by studying T cells reactive against viral peptides. We used a commercially available standardized set of 32 immunogenic viral epitopes (CEF peptides, derived from cytomegalovirus, EBV, and influenza). These epitopes are all restricted by frequently expressed HLA class Is and are commonly used as positive controls in functional studies of T-cell reactivity.22,23 PBMCs from 2 healthy HLA-A*02:01+ donors (donors 1 and 2) were stimulated with CEF peptides. On day 10, cultured cells were either restimulated with CEF peptide overnight or rested in media. CEF-reactive CD8+ T cells were isolated from the restimulated PBMCs by flow cytometry on the basis of IFNγ secretion (12.8% and 6.8% for donors 1 and 2, respectively; Figure 3A). We performed IFNγ ELISPOT assays against individual component peptides to deconvolute the antigen specificity of CEF pool-reactive T cells, focusing on peptides with HLA-restriction compatible with the HLA class I typing of the donors (supplemental Table 3B). Donor 1 (HLA-A*02:01, HLA-A*24:02, HLA-B*07:02) and donor 2 (HLA-A*02:01, HLA-A*01:01) had potential reactivity against 9 and 7 of 32 CEF peptides, respectively, with these HLA restrictions. ELISPOT results confirmed that donor 1 had T cells specific to 4 of these 9 peptides; namely, Influenza M158-66, EBV LMP2A426-434, EBV BMLF1259-267, and EBV EBNA3A379-387 (Figure 3B, top). Donor 2 had T cells specific for 5 of 7 peptides; namely, M158-66, LMP2A426-434, BMLF1259-267, Influenza PB1591-599, and Influenza NP44-52 (Figure 3B, bottom). To discover TCRs specific for CEF peptides, IFNγ+ CD8+ T cells from CEF-stimulated PBMCs were sorted for single-cell sequencing. We adapted a method of single-cell targeted TCR sequencing that employs primers specific for distinct Vα and Vβ regions to produce linked TCRα/TCRβ Illumina libraries (supplemental Table 4).4 In total, we obtained paired chain sequence information from 247 (67.1%) of 368 and 297 (80.7%) of 368 single cells for donors 1 and 2, respectively (Figure 3C; supplemental Table 5A-C). Compared with the CEF-nonreactive CD8+ T cells (no IFNγ production), the CEF-reactive CD8+ T cells showed evidence of clonal expansion. A total of 81 and 89 unique clonotypes were identified for donors 1 and 2, respectively, and the most dominant clone represented 22.3% (55 of 247, donor 1) and 33.0% (98 of 297, donor 2) of the isolated CEF-reactive T cells (Figure 3D). In both donors, we detected an enriched sequence that was previously reported as specific for the BMLF1259-267 epitope.24
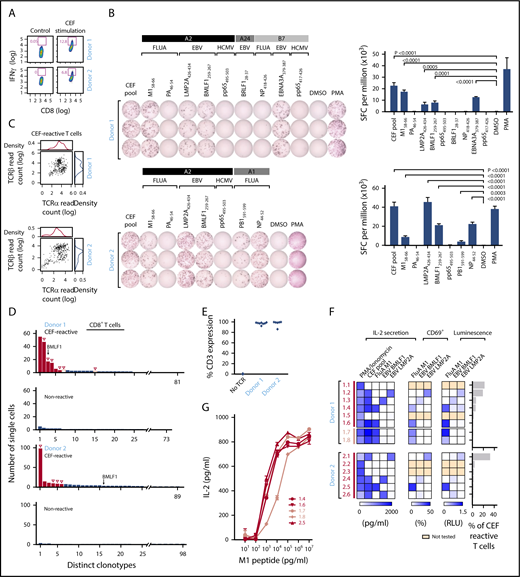
TCR sequences can be matched against pools of cognate viral peptides. (A) CEF-reactive CD8+ T cells were sorted by IFNγ secretion assay from 2 healthy donor (donor 1 and donor 2) PBMCs stimulated with the CEF peptides using flow cytometry. (B) Deconvolution of the reactivities of donor 1 and 2 PBMCs against individual peptides in the CEF pool using IFNγ ELISPOT. IFNγ secreting cells were quantified, as spot-forming cell (SFC) per million of CEF-reactive T cells (right; P values computed by comparison with dimethyl sulfoxide, using 1-sided 2-sample t-test of square root of spot counts). (C) Paired TCRα and TCRβ read counts from the sequencing of CEF-reactive single T cells. (D) Multiple paired TCRαβ sequences were enriched in CEF-reactive CD8+ T cells compared with nonreactive CD8+ T cells based on single-cell TCR sequencing (Red-selected for downstream cloning and expression; solid red bars, selected on the basis of prevalence; open red bars, selected based on TRBV usage; open triangles, TCRs for which antigen specificity was identified). A TCR with sequences identical to a BMLF1-specific TCR sequence previously reported in literature is indicated. (E) Dominant TCRs were expressed on Jurkat∆αβ reporter cells by lentiviral transduction, with stabilized expression of CD3 verified by flow cytometry. (F) Cognate antigens for CEF-reactive TCRs were determined by screening against individual peptides among the CEF peptide pool, with reactivity detected by IL-2 ELISA, CD69 staining, or luciferase assay. (G) Comparison of the functional avidity of FluA M1-specific TCRs identified from donors 1 and 2. EBV, Epstein-Barr virus; FluA, influenza A; HCMV, human cytomegalovirus.
TCR sequences can be matched against pools of cognate viral peptides. (A) CEF-reactive CD8+ T cells were sorted by IFNγ secretion assay from 2 healthy donor (donor 1 and donor 2) PBMCs stimulated with the CEF peptides using flow cytometry. (B) Deconvolution of the reactivities of donor 1 and 2 PBMCs against individual peptides in the CEF pool using IFNγ ELISPOT. IFNγ secreting cells were quantified, as spot-forming cell (SFC) per million of CEF-reactive T cells (right; P values computed by comparison with dimethyl sulfoxide, using 1-sided 2-sample t-test of square root of spot counts). (C) Paired TCRα and TCRβ read counts from the sequencing of CEF-reactive single T cells. (D) Multiple paired TCRαβ sequences were enriched in CEF-reactive CD8+ T cells compared with nonreactive CD8+ T cells based on single-cell TCR sequencing (Red-selected for downstream cloning and expression; solid red bars, selected on the basis of prevalence; open red bars, selected based on TRBV usage; open triangles, TCRs for which antigen specificity was identified). A TCR with sequences identical to a BMLF1-specific TCR sequence previously reported in literature is indicated. (E) Dominant TCRs were expressed on Jurkat∆αβ reporter cells by lentiviral transduction, with stabilized expression of CD3 verified by flow cytometry. (F) Cognate antigens for CEF-reactive TCRs were determined by screening against individual peptides among the CEF peptide pool, with reactivity detected by IL-2 ELISA, CD69 staining, or luciferase assay. (G) Comparison of the functional avidity of FluA M1-specific TCRs identified from donors 1 and 2. EBV, Epstein-Barr virus; FluA, influenza A; HCMV, human cytomegalovirus.
We cloned and expressed the 6 most dominant TCRs from each donor and transduced them into Jurkat∆αβ reporter cells. The transduction efficiency was 86% to 99.9%, determined by CD3 staining (Figure 3E). To interrogate the antigen specificity of each reconstructed TCR, we cocultured TCR-expressing reporter cells with APCs (K562-A*02:01 cells) pulsed with either the CEF pool or individual HLA-A*02:01+-restricted peptides that were shown to be reactive by ELISPOT. On the basis of IL-2 secretion, 4 of 6 TCRs from donor 1, and 4 of 6 TCRs from donor 2 were specific for a peptide in the pool. CEF-reactive TCRs were thereafter tested for reactivity against individual peptides. For donor 1, 2 of 4 TCRs were matched to M1, whereas 1 each was reactive to BMLF1 and LMP2A. Likewise, for donor 2, 2 of 4 TCRs matched to BMLF1, 1 to M1, and 1 to LMP2A. In all cases, similar patterns of TCR reactivity were observed by measuring upregulation of the T-cell activation marker CD69 on the reporter cells and by luciferase readouts (Figure 3F). The 4 unmatched TCRs could be specific for non-HLA-A*02:01-restricted CEF peptides or may have been nonspecifically selected by the IFNγ secretion assay. Two additional TCRs from donor 1 (D1.7 and D1.8) were chosen to be reconstructed on the basis of their TCRβ chain, TRBV19, which is highly prevalent in TCR repertoires against M1.25 These were also confirmed to be M1-specific. Consistent functional avidity across the 5 M1-specific TCRs from both donors was observed, with detection of reactivity to M1 peptide starting at the same peptide concentration, 1 ng/mL (Figure 3G).
Identification of neoantigen-specific TCRs after treatment with personalized neoantigen vaccines
The ability to assess functional avidity of discovered TCRs is highly relevant in the design of T-cell-based immunotherapies. We therefore applied our approach to study the T-cell response against neoantigens. There is growing evidence supporting the role of neoantigens in antitumor responses to immunotherapy.1,26 Recently, we reported the feasibility and immunogenicity of a personalized neoantigen-targeting vaccine for patients with high-risk melanoma.27 All enrolled patients had stage IIIB/C or IVM1a/b melanoma treated with surgical resection, and vaccine was administered as an adjuvant therapy to decrease risk for recurrence. In brief, neoantigen peptide vaccines were generated on the basis of whole-exome sequencing of tumor and matched normal cells to identify tumor-specific mutations and selection of neoantigens using NetMHCpan, the HLA class I-binding prediction algorithm.20 For each patient, up to 20 peptides (15-30 amino acids in length, containing the predicted epitope) as immunizing peptide (IMP) were administered with the adjuvant polyinosinic-polycytidylic acid stabilized with polylysine and carboxymethylcellulose (poly-ICLC) in a prime-boost schedule (Figure 4A). We previously reported the induction of circulating polyfunctional neoantigen-reactive CD4+ and CD8+ T cells after vaccination.27 We now asked whether we could identify TCRs specific for neoantigens targeted by vaccination and assess their ability to discriminate between mutant and wild-type peptide, focusing on 2 of the 6 study subjects (patients 1 and 3).

Neoantigen-specific TCR sequences are identified from patients with melanoma immunized with personal neoantigen vaccines. (A) TCR repertoire was assessed in PBMCs collected from melanoma patients (patients 1 and 3) ∼16 weeks after initiation of personalized neoantigen vaccination.27 (B) Neoantigen-reactive CD4+ and CD8+ T cells from patients 1 and 3, respectively, were isolated on the basis of IFNγ secretion after exposure of PBMCs to pools of 15 to 16 mer-length (CD4) and 9 to 10 mer-length (CD8) peptides corresponding to the immunizing peptides (*neoantigens previously demonstrated to be immunogenic based on bulk PBMC analysis). (C) Targeted single-cell TCR sequencing reveals multiple paired TCR sequences enriched in the neoantigen-reactive repertoires of patients 1 and 3. Red for downstream cloning and expression; open triangle for antigen-matched TCRs. (D) One of 6 reconstructed CD4+ TCRs from patient 1 was matched with mut-RUSC2-2 (mapping of this peptide within the original IMP indicated) per detection of reactivity by IL-2 ELISA. Functional avidity of the mut-RUSC2-2-specific P1.6 TCR against mutant and corresponding wild-type (WT) peptide was determined using autologous APCs (P = .0003, by 2-sample t-test with Welch’s correction from 4 technical replicates). (E) Three reconstructed CD8+ TCRs specific for mut-VPS16 were identified from patient 3 per screening by IL-2 ELISA, and mapping of the cognate peptide within the IMP is shown. The peptides to test against the TCRs were selected on the basis of reactivities against the peptides in bulk in vitro cultures (supplemental Figure 3). Functional avidity for mut- and WT-VPS16 by the P3.3, P3.6, and P3.7 TCRs was determined using B721.221 HLA-B*27:05 cell line pulsed with VPS16 peptides by IL-2 ELISA. Significant difference in IL-2 response was measured at 1 μg/mL mut-VPS16 peptide and corresponding WT peptide. (F) HLA-B*27:05 restriction of mut-VPS16 was verified by coculture of TCR-expressing reporter cells with peptide-pulsed K562 cells expressing HLA-A*02:01 or B721.221 cells expressing HLA-A*03:01 or HLA-B*27:05.
Neoantigen-specific TCR sequences are identified from patients with melanoma immunized with personal neoantigen vaccines. (A) TCR repertoire was assessed in PBMCs collected from melanoma patients (patients 1 and 3) ∼16 weeks after initiation of personalized neoantigen vaccination.27 (B) Neoantigen-reactive CD4+ and CD8+ T cells from patients 1 and 3, respectively, were isolated on the basis of IFNγ secretion after exposure of PBMCs to pools of 15 to 16 mer-length (CD4) and 9 to 10 mer-length (CD8) peptides corresponding to the immunizing peptides (*neoantigens previously demonstrated to be immunogenic based on bulk PBMC analysis). (C) Targeted single-cell TCR sequencing reveals multiple paired TCR sequences enriched in the neoantigen-reactive repertoires of patients 1 and 3. Red for downstream cloning and expression; open triangle for antigen-matched TCRs. (D) One of 6 reconstructed CD4+ TCRs from patient 1 was matched with mut-RUSC2-2 (mapping of this peptide within the original IMP indicated) per detection of reactivity by IL-2 ELISA. Functional avidity of the mut-RUSC2-2-specific P1.6 TCR against mutant and corresponding wild-type (WT) peptide was determined using autologous APCs (P = .0003, by 2-sample t-test with Welch’s correction from 4 technical replicates). (E) Three reconstructed CD8+ TCRs specific for mut-VPS16 were identified from patient 3 per screening by IL-2 ELISA, and mapping of the cognate peptide within the IMP is shown. The peptides to test against the TCRs were selected on the basis of reactivities against the peptides in bulk in vitro cultures (supplemental Figure 3). Functional avidity for mut- and WT-VPS16 by the P3.3, P3.6, and P3.7 TCRs was determined using B721.221 HLA-B*27:05 cell line pulsed with VPS16 peptides by IL-2 ELISA. Significant difference in IL-2 response was measured at 1 μg/mL mut-VPS16 peptide and corresponding WT peptide. (F) HLA-B*27:05 restriction of mut-VPS16 was verified by coculture of TCR-expressing reporter cells with peptide-pulsed K562 cells expressing HLA-A*02:01 or B721.221 cells expressing HLA-A*03:01 or HLA-B*27:05.
TCR characterization studies were performed using PBMCs obtained 1 month after the first boost (week 16). Our previous study showed that postvaccination, neoantigen-specific CD4+ T-cell responses were detectable ex vivo and neoantigen-specific CD8+ T-cell responses were detectable after 1 round of in vitro stimulation.27 For patient 1, 1 peptide pool (9 peptides total), containing overlapping 15 to 16 amino acid peptides encompassing 3 IMPs, stimulated CD4+ T-cell responses that we could detect ex vivo. On deconvolution, peptides derived from 2 IMPs (mut-CASP5, mut-RUSC2) were immunogenic ex vivo.27 Therefore, for patient 1, neoantigen-reactive CD4+ T cells were isolated from PBMCs ex vivo on the basis of IFNγ secretion after overnight stimulation with the pool (supplemental Table 3C). These IFNγ+ cells represented 0.521% of the CD4+ T-cell population after stimulation, which was almost 18-fold higher than the nonstimulated control population (0.03%; Figure 4B, top). For patient 3, PBMCs were stimulated in vitro for 21 days with a pool of peptides (12 peptides total, 9-10 amino acids derived from 7 IMPs, 3 of which were immunogenic [mut-CIT, mut-CASP1, and mut-VPS16]; supplemental Figure 3).27 CD8+ T cells were then isolated from these in vitro-stimulated PBMCs on the basis of IFNγ secretion after overnight stimulation. Here, the IFNγ+ cells represented 0.11% of the CD8+ T-cell population after pool stimulation, more than 3-fold higher compared with the control nonrestimulated cells (0.03%; Figure 4B, bottom). The pool-reactive T cells from both patients 1 and 3 were submitted for single-cell TCR sequencing (supplemental Table 5D-E). A total of 99 and 29 unique clonotypes of 106 and 119 cells sequenced were identified for patients 1 and 3, respectively, with the most dominant clone representing 2.8% (3 cells) and 25.2% (30 cells) of the isolated pool-reactive T cells, respectively (Figure 4C).
To probe the specificity of identified TCRs to neoantigens, 6 and 7 dominant TCRs from patients 1 and 3, respectively, were reconstructed and expressed. We screened for reactivity against peptides previously demonstrated to be immunogenic in each patient mentioned earlier. TCR-expressing reporter cells were cocultured with patient autologous APCs pulsed with individual immunogenic peptides in the pool. Mut-RUSC2 was found to be the cognate antigen for 1 TCR in patient 1 (P1.6), with clear discrimination between mutant and wild-type forms of peptide at 10 μg/mL (Figure 4D). Mut-VPS16, which was predicted to bind with high affinity to HLA-B*27:05, was found to be the cognate antigen for 3 TCRs in patient 3 (P3.3, P3.6, P3.7). All 3 mut-VPS16-specific TCRs discriminated between mutant and wild-type forms of the peptide, with recognition of mutant peptide detected at the same concentration thresholds (Figure 4E). We confirmed the HLA-B*27:05 restriction of this neopeptide by demonstrating IL-2 production selectively when the mutated peptide was pulsed on HLA-B*27:05-expressing cell lines, but not on the other patient-specific HLA-expressing cell lines (HLA-A*02:01 and HLA-A*03:01; Figure 4F).
Identification of TCRs specific for a novel predicted neoantigen in CLL
Antigen-specific T-cell-based therapies hold great promise, and their design requires knowledge of the precise TCRs corresponding to target antigens.28 To test whether our approach could be used for identifying TCR sequences specific for candidate novel neoantigens, we evaluated TCRs identified from HLA-matched healthy donor PBMCs stimulated against candidate CLL neoantigens. Neoantigens arising from insertion-deletions (indels) are of particular interest because the frameshift can result in a completely novel peptide sequence.29 To this end, we analyzed whole-exome sequencing data from 157 patients with CLL to identify predicted neoantigens arising from indels. One hundred twenty indels were identified from 80 patients, and of these, 28 indels were predicted to give rise to 76 neoantigens with half-maximal inhibitory concentration (IC50) below 500 nM (Figure 5A; supplemental Figure 4A-B). RNA sequencing or Sanger sequencing validated 13 of the 28 indel mutations. Seven validated indels were identified in HLA-A*02:01+ patients, with 12 candidate neopeptides, including 6 strong binders with predicted IC50 lower than 150 nM. These candidate epitopes were evaluated for experimental binding affinity to HLA-A*02:01, and the 5 with IC50 lower than 500 nM were tested for immunogenicity by IFNγ ELISPOT (supplemental Table 3D). HLA-A*02:01+ PBMCs from a healthy donor were stimulated with each neoantigen peptide weekly, for a total duration of 21 days. Two of the neoantigens (MGA_106 and ITPKB) were found to be immunogenic (supplemental Figure 4C), with the strongest response detected against mut-MGA_106. Mut-MGA_106 arises from a deletion mutation in MGA, a putative CLL driver, and has a predicted IC50 of 106.5 nM (Figure 5B). The mut-MGA response was abrogated by class I blocking antibody, confirming its HLA class I-restriction (Figure 5C).

Neoantigen-specific TCR sequences are identified for a newly characterized CLL neoantigen. (A) Summary of filtering process used to identify CLL neoantigens arising from indel mutations. (B) A frameshift deletion in a putative CLL driver MGA (4227 del T) generates a neoantigen, with predicted binding affinity of IC50 = 106.5 nM to HLA-A*02:01. Other mutations identified in 16 patients with CLL shown here were previously described (* denotes nonsense mutation).16 (C) IFNγ ELISPOT confirmed the immunogenicity of mut-MGA_106 peptide in HLA-A*02:01+ healthy donor PBMCs cultured with mut-MGA_106 peptide (P = .0029, compared with control peptide with 1-sided 2-sample t-test). Abrogation of response was observed with class I blocking antibody, W6/32 (P = .0005). (D) Single-cell TCR sequencing identified enriched clones (red for downstream cloning and expression; open triangle for matched mut-MGA_106-specific TCR). (E) Dominant TCRs were cloned and expressed on Jurkat∆αβ reporter cells, and cocultured with autologous APCs pulsed with mut-MGA_106 peptide. One mut-MGA-specific TCR was identified by IL-2 ELISA. (F) Functional avidity of mut-MGA-specific TCR is determined.
Neoantigen-specific TCR sequences are identified for a newly characterized CLL neoantigen. (A) Summary of filtering process used to identify CLL neoantigens arising from indel mutations. (B) A frameshift deletion in a putative CLL driver MGA (4227 del T) generates a neoantigen, with predicted binding affinity of IC50 = 106.5 nM to HLA-A*02:01. Other mutations identified in 16 patients with CLL shown here were previously described (* denotes nonsense mutation).16 (C) IFNγ ELISPOT confirmed the immunogenicity of mut-MGA_106 peptide in HLA-A*02:01+ healthy donor PBMCs cultured with mut-MGA_106 peptide (P = .0029, compared with control peptide with 1-sided 2-sample t-test). Abrogation of response was observed with class I blocking antibody, W6/32 (P = .0005). (D) Single-cell TCR sequencing identified enriched clones (red for downstream cloning and expression; open triangle for matched mut-MGA_106-specific TCR). (E) Dominant TCRs were cloned and expressed on Jurkat∆αβ reporter cells, and cocultured with autologous APCs pulsed with mut-MGA_106 peptide. One mut-MGA-specific TCR was identified by IL-2 ELISA. (F) Functional avidity of mut-MGA-specific TCR is determined.
To identify TCRs specific for mut-MGA, CD8+ T cells reactive to mut-MGA were isolated from donor PBMCs stimulated with mut-MGA peptide by flow cytometry and submitted for single-cell TCR sequencing. Of 31 mut-MGA-reactive CD8+ T cells, 23 unique clonotypes were identified, in which 3 clonotypes were identified more than once (Figure 5D; supplemental Table 5F). The top 3 clonotypes were reconstructed, and TCR-expressing reporter cells were cocultured with K562-A2 cells pulsed with either the mut-MGA or irrelevant peptide. Based on IL-2 production, 1 TCR was determined to be specific for mut-MGA (Figure 5E and 5F).
Discussion
The interaction between TCRs and antigens affects a wide range of disease, including cancer, infection, and autoimmune disorders.30,31 There is growing interest in developing methods to monitor antigen-specific T-cell responses and to identify TCR sequences capable of recognizing antigens of interest, given their potential for use as therapeutic agents.32,33
In this study, we developed a novel cloning and expression system to probe the specificity of TCRs discovered by single-cell sequencing. Key innovations include a variable chain plasmid library that allows any TCR of interest to be assembled by combining library components, with a custom oligonucleotide encoding the CDR3 sequences, and a TCR-deficient Jurkat cell line modified to express an NFAT-luciferase reporter, which allows for rapid screening of TCR activation. Generating TCR-expressing cell lines has several advantages over transduction of primary T cells, including the ability to expand and maintain a fixed repertoire of TCR-expressing cells indefinitely, and the absence of competition for expression of possible mispairing between chains of the transduced and endogenous TCR (although there are strategies to overcome this by using mouse constant regions).34 We applied this system to identify neoantigen-specific TCR sequences from patients with melanoma treated with personalized neoantigen vaccines and study TCR sequences specific for mut-MGA, a novel neoantigen identified in a patient with CLL. We were able to generate TCR-expressing reporter cells in 9 days after obtaining sequencing information, and we envision that our approach could be adapted for more high-throughput reconstruction of TCRs by incorporating pooled and automated cloning strategies.
A key advantage of our system is the ability to rapidly produce a homogenous population of T cells of defined TCR specificity, and to compare their functional avidity for a specific antigen of interest. Functional avidity is an essential parameter to consider in the design of effective T-cell-based immunotherapies, as T cells with higher value may mediate superior antitumor responses, although they may also become more readily exhausted because of extended antigen activation.35,36 Functional avidity is dependent on many factors, including the level of TCR expression on the cell, the density of MHC molecules binding a specific peptide on the APC surface, the extent of co-aggregation of costimulation molecules with the TCR, and the state of the T cell (ie, naive or memory).37 Our reporter cells provide a controlled system, with many of these variables fixed or tunable, such that TCRs specific to an antigen of interest can be directly compared. Using this approach, we found that the 4 neoantigen-specific TCRs identified in patients treated with neoantigen-based vaccine had relatively lower functional avidity compared with our previous bulk T cells,27 but were yet able to discriminate between the mutant and wild-type forms of the epitope. These differences are likely because the reporter cells are expressing TCRs from a single T-cell clone, whereas the primary T-cell lines27 contain multiple T-cell clones with a diversity of TCRs. We expect vaccination to stimulate T cells with a range of functional avidities, but the TCRs that we have cloned for this proof-of-concept study are restricted to only a small subset. Thus, although these results demonstrate the specificity of reconstituted TCRs for the tumor neoantigens, they also highlight the challenges still to overcome for complete dissection of immune responses after therapy, which will require the ability to perform a larger-scale evaluation of a substantially larger set of TCRs.
In addition to revealing insights about TCR function, this method could also provide a means to improve selection of immunogenic epitopes, and in turn optimize cancer vaccine design. Current neoantigen prediction algorithms center primarily on the peptide-MHC binding interaction, but remain imperfect predictors of immunogenicity.19,38,39 We provide a means to systematically study the relationship between predicted immunogenicity and observed TCR avidity, which could improve our understanding of the characteristics of immunogenic epitopes and generate data to train algorithms to improve neoantigen prediction.
A further challenge in the study of TCRs is deciphering T-cell specificity based on primary sequence data alone when there is no predetermined pool of candidate antigens. Indeed, the cognate antigens of a number of TCRs reconstructed in our study were not identified as among the pool of peptides tested, despite the fact that their selection for single-cell TCR sequencing was based on reactivity against these peptide pools. A likely reason for this is related to nonspecific binding of the capture reagent used to isolate IFNγ-secreting T cells in the stimulation culture. Another possibility, although less likely, is that an incorrect TCRαβ pairing was reconstructed (we consistently reconstituted the most dominant pair), because of the presence of more than 1 TCRα in the cell4,40 ; we cannot exclude the possibility that a less dominant TCRα paired with the TCRβ chain to create a functional TCR. Further investigation will be required to definitively establish the antigen specificity of those TCRs beyond those strictly reactive to the expected set of test peptides.
Emerging algorithms that define TCR motifs associated with antigen specificity are making strides toward deciphering the rules of TCR-antigen recognition.24,41 Our pipeline could be used with such algorithms, facilitating investigations into the specificity of TCRs clustered according to sequence similarity, and expediting the search for cognate antigens. Together, these applications hold great promise for improving immune monitoring, and guiding the design of effective T-cell based immunotherapies.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the Dana-Farber Cancer Institute (DFCI) Pasquarello Tissue Bank and the Broad Institute’s Biological Samples, Genetic Analysis, and Genome Sequencing Platforms. The authors are also grateful to Giacomo Oliveira and Pavan Bachireddy for helpful discussions.
This research was made possible by generous gifts from the Blavatnik Family Foundation and the G. Harold and Leila Y. Mathers Charitable Foundation, and was supported by grants from the Broad Institute Scientific Projects to Accelerate Research and Collaboration program, Broad Next10 Seed funding, and the National Institutes of Health, National Cancer Institute (1RO1CA155010-02) and National Institutes of Health, National Heart, Lung, and Blood Institute (5R01HL103532-03). This work was further supported by a DFCI Center for Cancer Immunotherapy Research fellowship (Z.H.), National Institutes of Health, National Cancer Institute T32CA207021 (Z.H.), Howard Hughes Medical Institute Medical Research Fellowship (A.J.A.), American Society of Hematology HONORS Award (A.J.A.), PhRMA Fellowship for Translational Medicine and Therapeutics (J. Sun), National Institutes of Health, National Cancer Institute R50RCA211482A (S.A.S.), National Institutes of Health, National Cancer Institute R21 CA216772 and National Cancer Institute–Specialized Programs of Research Excellence (SPORE) 2P50CA10194211A1 (D.B.K.). C.J.W. is a Scholar of the Leukemia and Lymphoma Society.
Authorship
Contribution: Z.H. and A.J.A. designed and performed experimental and data analysis together with J.K., D.E.L., D.J.B., C.C., L.W., W.Z., D.T., S.S., K.J.L., U.E.B., and D.B.K; U.E.B. and E.F.F. designed the library cloning strategy; J. Sun and S.A.S. performed computational analysis; I.G., J. Stevens, and W.J.L. performed HLA typing; H.K. and A.M. generated the TCR-deficient Jurkat cell line; D.N. performed statistical analysis; E.F.F., P.A.O., N.H., and C.J.W. directed the overall NeoVax study design; O.O. coordinated clinical research; Z.H., A.J.A., J. Sun, and C.J.W. wrote the manuscript; and all authors discussed and interpreted results.
Conflict-of-interest disclosure: S.A.S. has equity in 152 Therapeutics; E.F.F. is a cofounder and employee of Neon Therapeutics; N.H. and C.J.W. are cofounders of Neon Therapeutics and members of its scientific advisory board. As a result of this interest, C.J.W. is subject to a conflict-of-interest management plan, which prohibited her from participating in the referenced clinical trial of vaccination with a personal neoantigen-targeting melanoma vaccine (NCT01970358). P.A.O. has advised Neon Therapeutics. The remaining authors declare no competing financial interests.
The current affiliation for U.E.B. is Accelerating Cancer Immunotherapy Research, Cambridge, MA.
The current affiliation for E.F.F. is Neon Therapeutics, Cambridge, MA.
Correspondence: Catherine J. Wu, Dana-Farber Cancer Institute, Dana 520, 450 Brookline Ave, Boston MA 02215; e-mail: cwu@partners.org.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal