

Abstract
With the advent of tyrosine kinase inhibitors (TKIs), the goals of therapy in chronic myeloid leukemia (CML) are steadily shifting. Long-term disease control on TKI therapy has been the goal and expectation for most patients. More recently, treatment-free remission (TFR) has entered mainstream practice and is increasingly being adopted as the main goal of therapy. This therapeutic shift not only influences TKI selection but also, has necessitated the refinement and dissemination of highly sensitive and accurate molecular monitoring techniques. Measurement of BCR-ABL1 messenger RNA expression through reverse transcription quantitative polymerase chain reaction, reported according to the International Scale, has become the primary tool for response assessment in CML. Achieving specific time-dependent molecular milestones, as defined by global therapeutic guidelines, has been established as critical in maximizing optimal outcomes while identifying patients at risk of therapy failure. Depth and duration of a deep molecular response have become the new therapeutic targets in patients considered for TFR. Consequently, molecular monitoring in CML has become even more critical to ongoing response assessment, identifying patients with TKI resistance and poor drug adherence, and enabling TFR to be attempted safely and effectively.
What constitutes molecular monitoring?
After achievement of a complete cytogenetic response (CCyR), equating to a BCR-ABL1 reverse transcription quantitative polymerase chain reaction (RT-qPCR) of ∼1% (International Scale [IS]), patient monitoring via conventional bone marrow metaphase analysis has minimal value unless CCyR is lost. Qualitative polymerase chain reaction (PCR) has limited value except at diagnosis, where it should be used to identify patients with atypical BCR-ABL1 transcripts. Quantitative PCR assays of peripheral blood, measuring the level of BCR-ABL1 messenger RNA relative to an internal control gene, are the gold standard for monitoring in chronic myeloid leukemia (CML), with the degree of leukemic cell burden translating to critically important prognostic information. Real time RT-qPCR is the primary tool for monitoring CML response and has the advantages of reliability, sensitivity, ease of replicability, and relatively rapid results. Other quantitative strategies previously used include nested PCR, which is more sensitive than RT-qPCR but more labor intensive, requiring days to generate a result.1 Multiplex PCR is useful at diagnosis to identify the BCR-ABL breakpoint, enabling atypical transcript detection.2 More novel techniques, such as digital and DNA-based PCR, can improve testing sensitivity, but they are not widely available and are currently research tools only.3,4 Establishment of the concordance of BCR-ABL1 RT-qPCR measurements in blood with bone marrow cytogenetics assessment5 and BCR-ABL1 values6 has reduced the requirement for invasive testing, whereas development of the IS has allowed for harmonization of testing between individual institutions.7
On the IS, patient results are expressed relative to the standardized baseline of 100% (for example, 0.1% BCR-ABL1 [IS] is a 3-log reduction equating to a major molecular response [MMR]).7 IS reporting by individual laboratories of BCR-ABL1 RT-qPCR is dependent on a predetermined conversion factor, generally derived from sample exchange with reference laboratories. Although IS development removed many variables leading to interlaboratory discrepancy, other factors exist. Utility of different control genes (ie, ABL vs GUSB) may influence the consistency of the conversion factor at high BCR-ABL transcript levels.7 The sample quality is crucial in ensuring accuracy, and specimens should be processed rapidly to minimize RNA degradation.8 Appropriate quality assurance should be undertaken to maximize accuracy and precision, whereas testing should be performed in replicate to minimize error.8
Case 1
A 52-year-old man presented with marked leucocytosis with associated neutrophilia, eosinophilia, and basophilia with a myelocyte peak. Bone marrow biopsy confirmed chronic phase (CP) CML, with karyotype analysis showing the Philadelphia chromosome. Molecular studies of peripheral blood revealed a BCR-ABL1 RT-qPCR result of 90% (IS). Sokal score was high, and he was commenced on nilotinib therapy at 300 mg twice daily. Complete hematological response was achieved within 6 weeks of therapy. BCR-ABL1 values at 1 and 3 months after nilotinib commencement were 80% and 25%, respectively. Is this response adequate? If not, should this patient be considered for a switch in tyrosine kinase inhibitor (TKI) therapy?
Early responses (0-18 months)
Response to TKI therapy is an important prognostic measurement, irrespective of which TKI is commenced in the first-line setting. The importance of these time-dependent milestones (Table 1) has been highlighted by several clinical trials and incorporated into global guidelines, such as those published by the European LeukemiaNet (ELN),9 the National Comprehensive Cancer Network (NCCN)10 and the European Society of Medical Oncology (ESMO).11 Response criteria have been divided into 3 categories, with “optimal” responses associated with the best long-term outcomes. Patients falling into the “failure” category should, with rare exception, switch therapy to minimize risk of disease progression and death. However, in some cases, a switch may not be appropriate (for instance, if poor adherence or drug interruption has contributed to the poor response), although differentiating noncompliance from TKI resistance may not be straightforward. Mutation analysis should be performed in “failure” patients, because BCR-ABL1 kinase domain (KD) mutations are the primary known cause of TKI resistance.12 However, controversy remains about patients falling into the “warning” category, previously termed “suboptimal,” particular those falling into the warning category at early time points.
| Month | Optimal response | Warning | Failure |
|---|---|---|---|
| 3 mo | |||
| ELN | BCR-ABL1 ≤ 10% ± Ph+ ≤ 35% | BCR-ABL1 > 10% ± Ph+ 36%-95% | No CHR or Ph+ > 95% |
| NCCN | BCR-ABL1 ≤ 10% | BCR-ABL1 > 10% | NA |
| ESMO | BCR-ABL1 < 10% ± Ph+ ≤ 35% | BCR-ABL1 > 10% ± Ph+ 36%-95% | No CHR or Ph+ > 95% |
| 6 mo | |||
| ELN | BCR-ABL1 < 1% ± Ph+ 0 | BCR-ABL1 1%-10% ± Ph+ 1%-35% | BCR-ABL1 > 10% ± Ph+ > 35% |
| NCCN | BCR-ABL1 ≤ 10% | NA | BCR-ABL1 > 10% |
| ESMO | BCR-ABL1 < 1% ± Ph+ 0% | BCR-ABL1 1%-10% ± Ph+ 1%-35% | BCR-ABL1 > 10% or Ph+ > 35% |
| 12 mo | |||
| ELN | BCR-ABL1 ≤ 0.1% | BCR-ABL1 > 0.1%-1% | BCR-ABL1 > 1% ± Ph+ > 0 |
| NCCN | BCR-ABL1 < 1% | BCR-ABL1 1%-10% | BCR-ABL1 > 10% |
| ESMO | BCR-ABL1 < 0.1% | BCR-ABL1 0.1%-1% | BCR-ABL1 > 1% ± Ph+ ≥ 1 |
| 18 mo | |||
| ESMO | BCR-ABL1 < 0.01% | BCR-ABL1 0.1%-1% | NA |
| At any time >12 mo | |||
| ELN | BCR-ABL1 ≤ 0.1% | CCA/Ph− (-7 or 7q-) | Loss of CHR |
| Loss of CCyR | |||
| Confirmed loss of MMR* | |||
| Mutations | |||
| CCA/Ph+ | |||
| NCCN | BCR-ABL1 < 0.1% | BCR-ABL1 0.1%-1% | BCR-ABL1 > 1% |
| Clinical considerations9,10 | Monitor response and side effects | Evaluate for patient compliance and drug interactions | Evaluate for patient compliance and drug interactions |
| No change to patient therapy | Consider mutation analysis | Consider mutation analysis | |
| Recommend TKI switch if no compliance issues or drug interactions identified |
| Month | Optimal response | Warning | Failure |
|---|---|---|---|
| 3 mo | |||
| ELN | BCR-ABL1 ≤ 10% ± Ph+ ≤ 35% | BCR-ABL1 > 10% ± Ph+ 36%-95% | No CHR or Ph+ > 95% |
| NCCN | BCR-ABL1 ≤ 10% | BCR-ABL1 > 10% | NA |
| ESMO | BCR-ABL1 < 10% ± Ph+ ≤ 35% | BCR-ABL1 > 10% ± Ph+ 36%-95% | No CHR or Ph+ > 95% |
| 6 mo | |||
| ELN | BCR-ABL1 < 1% ± Ph+ 0 | BCR-ABL1 1%-10% ± Ph+ 1%-35% | BCR-ABL1 > 10% ± Ph+ > 35% |
| NCCN | BCR-ABL1 ≤ 10% | NA | BCR-ABL1 > 10% |
| ESMO | BCR-ABL1 < 1% ± Ph+ 0% | BCR-ABL1 1%-10% ± Ph+ 1%-35% | BCR-ABL1 > 10% or Ph+ > 35% |
| 12 mo | |||
| ELN | BCR-ABL1 ≤ 0.1% | BCR-ABL1 > 0.1%-1% | BCR-ABL1 > 1% ± Ph+ > 0 |
| NCCN | BCR-ABL1 < 1% | BCR-ABL1 1%-10% | BCR-ABL1 > 10% |
| ESMO | BCR-ABL1 < 0.1% | BCR-ABL1 0.1%-1% | BCR-ABL1 > 1% ± Ph+ ≥ 1 |
| 18 mo | |||
| ESMO | BCR-ABL1 < 0.01% | BCR-ABL1 0.1%-1% | NA |
| At any time >12 mo | |||
| ELN | BCR-ABL1 ≤ 0.1% | CCA/Ph− (-7 or 7q-) | Loss of CHR |
| Loss of CCyR | |||
| Confirmed loss of MMR* | |||
| Mutations | |||
| CCA/Ph+ | |||
| NCCN | BCR-ABL1 < 0.1% | BCR-ABL1 0.1%-1% | BCR-ABL1 > 1% |
| Clinical considerations9,10 | Monitor response and side effects | Evaluate for patient compliance and drug interactions | Evaluate for patient compliance and drug interactions |
| No change to patient therapy | Consider mutation analysis | Consider mutation analysis | |
| Recommend TKI switch if no compliance issues or drug interactions identified |
All BCR-ABL1 values are expressed using IS. CCA/Ph−, clonal chromosome abnormalities in Ph− cells; CCA/Ph+, clonal chromosome abnormalities in Ph+ cells; CHR, complete hematological response; ELN, European LeukemiaNet; ESMO, European Society of Medical Oncology; NA, not applicable; NCCN, National Comprehensive Cancer Network; Ph+, Philadelphia-positive cells.
In 2 consecutive tests, one had BCR-ABL1 transcripts ≥1%.
Early molecular response
Differentiation of patients with poor outcomes from the majority with optimal responses has been a major area of research. The prognostic implication of early predictors, such as early molecular response (EMR), which is traditionally defined as BCR-ABL1 ≤ 10% (IS) 3 months after TKI commencement, has been shown by numerous trials. This molecular threshold, developed in parallel by Hanfstein et al13 and Marin et al,14 was established as being predictive of superior overall survival (OS), progression-free survival (PFS), CCyR, and molecular responses in patients treated with frontline imatinib. As a predictor for future eligibility for treatment-free remission (TFR), patients who fail to achieve EMR have the poorest achievement of stable MR4.5 (BCR-ABL1 ≤ 0.0032% [IS]) after 8 years of imatinib therapy in an Australian series of 423 patients (2 of 84 patients; 2%), highlighting the link between EMR and TFR eligibility.15
The value of EMR has also been established in patients treated with second generation TKIs, with data from the 5-year update from the ENESTnd study revealing that EMR achievement was associated with superior rates of MR4.5 at 5 years compared with those with EMR failure.16 Survival measurements (OS and PFS) were also superior in patients successfully achieving EMR.16 Analysis of the dasatinib cohort of the SPIRIT 2 study showed that patients failing to achieve EMR had significantly poorer achievement of CCyR, MMR, and MR4.5 by 24 months.14 In the 5-year update of the DASISION study, patients achieving EMR had a significantly higher likelihood of reaching molecular targets, including MR4.5, with lower transformation rates and superior survival outcomes.17
However, although achieving EMR clearly is an important predictor of outcome, insufficient evidence exists to mandate a switch of therapy based on this single result, because some patients who fail to achieve EMR still reach optimal outcomes.18 The NCCN recommends interpreting the 3-month BCR-ABL1 value with caution in patients with values only slightly >10%, especially if there has been a rapid decline from baseline.10 Combining the 3- and 6-month disease reassessments may further assist in differentiating patients,19,20 but it may also delay an effective switch to more potent therapy in some cases.
Another differentiator for patients failing to achieve EMR is the kinetics of BCR-ABL1 decline. Branford et al18 described that a halving time of <76 days at 3 months in patients not achieving EMR was associated with superior outcomes compared with those with slower rates of decline (OS: 95% vs 58%, respectively; PFS: 92% vs 63%, respectively; MMR 54% vs 5%, respectively). A time-specific differentiator was not identified in the CML IV study, but OS and PFS were superior in patients with a minimum of half-log reduction in BCR-ABL1 values between baseline and 3 months.21 Although EMR remains an important early landmark for clinicians, there is a paucity of consensus agreement regarding TKI switch in the event of EMR failure.
Case 2
A 34-year-old woman diagnosed with CML 6 years ago is considering pregnancy. Her Sokal score at diagnosis was low, and she was treated with first-line imatinib 400 mg daily with reasonable tolerance. She achieved and maintained MMR after 18 months of imatinib but has not yet been able to achieve sustained deeper responses, despite excellent medication compliance. She would like to discuss TFR and her likelihood of achieving this. What is your advice?
Timing of MMR achievement
Although the ELN and the ESMO have classified MMR achievement at 12 months as optimal, the NCCN diverges from this recommendation by accepting a molecular response <1% at this time point. Conflicting data contribute to the lack of clarity regarding the timing of MMR achievement. Long-term follow-up from the IRIS study determined that imatinib-treated patients with MMR achievement at 12 months had superior 10-year OS (91.1% vs 85.3%) with fewer CML-related deaths (97.8% vs 89.4%) compared with those failing this target.22 Similarly, the 10-year follow-up of the CML IV study showed that MMR achievement at 12 months had statistically significant positive differences in OS, PFS, MR4 (BCR-ABL1 ≤ 0.01% [IS]), and MR4.5 attainment.23
Although the 3-year follow-up of the DASISION study showed that patients with MMR or CCyR achievement at 12 months had significantly better PFS compared with those who did not, this was not statistically significant.24 Furthermore, achievement of CCyR at 12 months was predictive of OS irrespective of MMR status.24 Similar findings were established by Marin et al25 in a cohort of 224 CP CML patients treated at a single institution; CCyR achievement at 12 months conferred a benefit in 5-year OS and PFS, whereas MMR failed to show any statistically significant difference. Evaluation by Jabbour et al26 of 167 patients treated with second generation TKIs in CP CML revealed that event-free survival was similar in patients achieving CCyR by 12 months, regardless of molecular response. Additional analysis of the CML IV study cohort validated the landmark of 12 to 15 months for achievement of a BCR-ABL1 value of 1%, whereas therapy failure should be considered if patients have failed to achieve MMR by a cutoff of 2.5 years, perhaps providing a deadline for when to consider TKI switch for patients persistently in the warning zone.27
Although it is unclear whether MMR achievement by 12 months affects survival, early achievement of MMR does predict subsequent achievement of MR4.5, important for treatment discontinuation consideration. Utilizing data from the CML IV study, Hehlmann et al28 showed that MMR achievement at 3, 6, 12, and 18 months had 5-year cumulative incidence rates of MR4.5 of 83.3%, 69.2%, 56.7%, and 51.1%, respectively (Table 2). Comparatively, a BCR-ABL1 value of 0.1% to 1% at 12 and 18 months had 5-year cumulative incidence rates of MR4.5 of only 14.5% and 6%, respectively.28 Similar results were observed by the Australian group, emphasizing the relationship between the rate of MR4.5 and early MMR achievement (Table 2).15 Early MMR achievement was also conducive to maintaining stable MR4.5 (ie, sustained MR4.5 for 2 years), which is important for TFR success.15
Cumulative incidence of MR4.5 depending on time point–specific molecular responses
Frequency of molecular monitoring
Although BCR-ABL1 RT-qPCR is not necessarily mandated at baseline, because subsequent quantification is aligned with the IS, the rate of BCR-ABL1 decline is useful for prognostication.18,21 Consequently, we recommend BCR-ABL1 quantification at baseline for all patients. After TKI initiation, 3-month BCR-ABL1 RT-qPCR testing is generally recommended until stable MMR is achieved followed by 3- to 6-month testing thereafter.9,11 More frequent molecular monitoring (3-4 tests per year) has been associated with increased TKI compliance, suggesting that 3-month BCR-ABL1 testing might encourage improved medication adherence.29 Our recommendation is to maintain a 3-month testing schedule where possible, especially in select populations: (1) patients with compliance concerns despite the achievement of MMR, (2) patients being considered for future TFR attempts to ensure sustained deep molecular response (DMR), and (3) patients switching to a new TKI. We recommend monthly monitoring in specific scenarios: (1) female patients who have ceased TKI for pregnancy, (2) patients entering a TFR attempt for the first 6 months after TKI discontinuation, and (3) patients attempting TFR who are beyond 6 months and have lost MR4.5 but maintain MMR.
Transcript type
The transcript generated from the BCR-ABL fusion gene is dependent on the site of breakage in BCR. Numerous studies have attempted to differentiate the effect of the common transcripts (b2a2 or e13a2; b3a2 or e14a2) on long-term outcomes. Although patients with b3a2 transcripts (alone or in combination with b2a2) achieve ELN-defined milestones more rapidly and have superior event-free survival,30 patients with b2a2 still have excellent outcomes. Although the majority of patients have common transcripts, ∼5% have atypical transcripts, which have implications for monitoring and long-term therapeutic decisions.2 The majority of laboratories cannot quantify atypical transcripts, and therefore, response assessments based on the prespecified milestones are difficult to apply. Cytogenetic assessments are useful until CCyR is achieved, whereas at subsequent time points, qualitative PCR may be used. By comparing the intensity of the PCR amplicon on gel electrophoresis with the sample’s low-level controls, an approximate value may be provided. TFR should generally be avoided in this setting due to an inability to accurately monitor transcript levels.
Mutation analysis
KD mutations are the best-described mechanism of TKI resistance, and mutation screening should be performed in patients failing TKI therapy. Detection of a KD mutation should direct subsequent TKI selection based on known resistance patterns to available TKIs.31,32 Failure to achieve milestones or loss of response should trigger investigation for mutations. Direct (Sanger) sequencing is the currently accepted technique for investigation of KD mutations, but it has relatively low sensitivity and can only detect mutations present in samples with ≥10% to 20% Philadelphia-positive cells, assuming that the BCR-ABL1 value is ≥1%.33 This level of detection is generally adequate in characterizing the relevant mutation due to the rapid expansion of the resistant clone, leading to a rise in BCR-ABL1 transcripts. Mutation analysis should be performed before stopping the TKI that has failed to maximize detection success. This is because, after the TKI is stopped, mutations can rapidly become difficult to detect. More sensitive techniques, such as mass spectrometry, digital droplet PCR, and next generation sequencing, can improve the detection limit to ∼0.2%, but these techniques are not widely available.33 Presence of multiple low-level mutations in patients with proven TKI failure has been predictive of poorer outcomes in the setting of inappropriate TKI selection.34,35 However, the significance of low-level mutations at diagnosis or in the absence of suspected resistance is unknown, and testing is not indicated outside of a clinical trial. KD domain mutations are only identified in about one-half of the patients with imatinib failure, and patients do not always respond to the appropriately selected agent, indicating the presence of other resistance pathways in many cases.
DMR
The duration and depth of molecular response have become synonymous with TFR eligibility. The term “complete molecular response,” implying undetectable BCR-ABL1 transcripts, has largely been abandoned due to significant variability in assay and sample sensitivity. “DMR” is the preferred expression, and the various definitions used to represent DMR describe the depth of response relative to the standardized baseline (eg, MR4 is ≥4-log reduction or ≤0.01% BCR-ABL1).4
Selection of more potent kinase inhibitors upfront can accelerate achievement of DMR and increase the percentage of patients who achieve it. The 5-year follow-up of the ENESTnd study showed that 31% of the imatinib-treated group achieved MR4.5, whereas 54% and 52% of the 2 nilotinib dosing arms reached MR4.5 by this time point.16 Similar results were observed at the 5-year update of the DASISION study, where dasatinib treatment was associated with higher rates of MR4.5 compared with imatinib (42% vs 33%).17 High-dose imatinib (ie, 800 mg daily as opposed to 400 mg daily) shows comparable rates of MR4.5 with second generation TKIs, although the higher dose is associated with more adverse events and rarely used today.36 Regardless, TKI selection for DMR achievement should be tempered with toxicity consideration, because each drug has a relatively unique adverse effect profile. Aiming for MMR or CCyR as opposed to DMR in patients who do not regard TFR as a high priority (ie, the elderly and patients with significant comorbidities) is appropriate, and therapy selection can be tailored to reflect this.
Switching to second-line therapy due to failure to achieve DMR with frontline imatinib has been proven to be a successful strategy. The 48-month analysis of the ENESTcmr study showed that, for patients treated with long-term imatinib with detectable molecular disease despite reaching CCyR, switching to nilotinib significantly increased the likelihood of MR4.5 achievement.37 For imatinib-treated patients in whom the best response has been CCyR but not MMR, switching to nilotinib can also induce MR4.5, whereas patients randomized to ongoing imatinib had little prospect of achieving MR4.5.37 Although robust data are not yet available for second-line dasatinib, switching to dasatinib after imatinib intolerance or failure allowed for MR4 achievement and eventual successful TFR attempts.38
Case 3
A 50-year-old man who was being treated for CML with upfront imatinib at 400 mg daily for the past 14 years stopped imatinib as part of a supervised TFR attempt. At diagnosis, his Sokal risk score was intermediate. He achieved EMR by 3 months and MMR by 12 months. He struggled with severe fatigue and gastrointestinal toxicity secondary to imatinib, which was a significant motivator for the TFR attempt. He eventually achieved MR4.5 after 12 years. After maintaining MR4.5 for 24 months, he ceased imatinib in a TFR attempt. Within 3 months, he had lost MMR, requiring TKI restart. By the time that he restarted imatinib, he had lost CCyR. CCyR was rapidly regained after imatinib recommencement, but he had not yet achieved MMR after 6 months of treatment. Is this an appropriate response? What should your approach be?
TFR
Several trials have shown the safety and viability of TFR in selected patients, and TFR is now incorporated into global guidelines (Table 3).9,10 TFR success has a number of health and economic implications, including eliminating the morbidity associated with TKI toxicity and a reduction in therapy costs for the patient, the drug funder, or both. The potential savings is illustrated by the drug costs saved in the Euro-Ski study, which were estimated at €22 million.39 Successful TFR has been achieved in ∼40% to 60% of patients enrolled in the various stopping studies, and most patients rapidly regain molecular control if TKI recommencement is required. Achieving and maintaining DMR are currently the fundamental requirements for TFR eligibility. The French STIM1 study, the first of the 2 pivotal stopping studies, ceased imatinib in 100 patients in sustained MR5 for ≥2 years, with the trigger to TKI recommencement being ≥1-log increase in BCR-ABL1 transcripts or loss of MMR. At 6 months, the rate of TFR was 43%, but this fell to 38% by 60 months.40 Similar results were observed in the Australian TWISTER study, where the entry criterion was ≥2 years of undetectable BCR-ABL transcripts (comparable retreatment criteria with the STIM1 study); the 24-month successful TFR rate was 47%.41
Summary of selected larger clinical trials of TFR relative to depth and duration of DMR
| Clinical study | Patient no. | TKI at time of cessation | Depth of DMR | Duration of DMR | TKI restart criteria | TFR success (follow-up) |
|---|---|---|---|---|---|---|
| STIM140 | 100 | Imatinib | UMRD | ≥2 yr | Loss of MMR or ≥1-log increase in BCR-ABL1 | 38% (60 mo) |
| TWISTER41 | 40 | Imatinib | UMRD | ≥2 yr | Loss of MMR or rising BCR-ABL1 on 2 consecutive tests | 47.1% (24 mo) |
| KID49 | 90 | Imatinib | UMRD | ≥2 yr | Loss of UMRD on 2 consecutive tests of MMR loss | 58.5% (24 mo) |
| STOP 2G-TKI44 | 60 | Dasatinib or nilotinib | UMRD | ≥2 yr | Loss of MMR | 53.57% (48 mo) |
| A-STIM42 | 80 | Imatinib | UMRD (occasional weakly positive samples also eligible) | ≥2 yr | Loss of MMR | 61% (36 mo) |
| ENESTFreedom43 | 190 | First-line nilotinib | MR4.5 | ≥1 yr | Loss of MMR | 51.6% (48 wk) |
| ENESTop47 | 126 | Second-line nilotinib after imatinib | MR4.5 | ≥1 yr | Loss of MMR or confirmed loss of MR4 | 53% (96 wk) |
| ISAV3 | 112 | Imatinib | MR4-MR4.5 | ≥18 mo | Loss of MMR | 48.1% (21.6 mo) |
| DADI38 | 63 | Second-line dasatinib | MR4 or 0.0069% (IS) | ≥1 yr | Loss of DMR | 44% (36 mo) |
| D-STOP50 | 54 | Dasatinib | MR4 | ≥1 yr | BCR-ABL1 > 0.0069% on 2 consecutive results | 57% (24 mo) |
| EURO-SKI39 | 755 | Imatinib > nilotinib/dasatinib | MR4 | ≥1 yr | Loss of MMR | 50% (24 mo) |
| Interim results | ||||||
| STIM2 (interim)53 | 124 | Imatinib | UMRD | ≥2 yr | Loss of MMR or ≥1-log increase in BCR-ABL1 | 59% (12 mo) |
| DASFREE (interim)54 | 84 | Dasatinib (first or second line) | MR4.5 | ≥1 yr | Loss of MMR | 49% (12 mo) |
| DESTINY55 | 117 | Imatinib > dasatinib/nilotinib | MR4 | ≥1 yr | Loss of MMR | 77% (24 mo) |
| Clinical study | Patient no. | TKI at time of cessation | Depth of DMR | Duration of DMR | TKI restart criteria | TFR success (follow-up) |
|---|---|---|---|---|---|---|
| STIM140 | 100 | Imatinib | UMRD | ≥2 yr | Loss of MMR or ≥1-log increase in BCR-ABL1 | 38% (60 mo) |
| TWISTER41 | 40 | Imatinib | UMRD | ≥2 yr | Loss of MMR or rising BCR-ABL1 on 2 consecutive tests | 47.1% (24 mo) |
| KID49 | 90 | Imatinib | UMRD | ≥2 yr | Loss of UMRD on 2 consecutive tests of MMR loss | 58.5% (24 mo) |
| STOP 2G-TKI44 | 60 | Dasatinib or nilotinib | UMRD | ≥2 yr | Loss of MMR | 53.57% (48 mo) |
| A-STIM42 | 80 | Imatinib | UMRD (occasional weakly positive samples also eligible) | ≥2 yr | Loss of MMR | 61% (36 mo) |
| ENESTFreedom43 | 190 | First-line nilotinib | MR4.5 | ≥1 yr | Loss of MMR | 51.6% (48 wk) |
| ENESTop47 | 126 | Second-line nilotinib after imatinib | MR4.5 | ≥1 yr | Loss of MMR or confirmed loss of MR4 | 53% (96 wk) |
| ISAV3 | 112 | Imatinib | MR4-MR4.5 | ≥18 mo | Loss of MMR | 48.1% (21.6 mo) |
| DADI38 | 63 | Second-line dasatinib | MR4 or 0.0069% (IS) | ≥1 yr | Loss of DMR | 44% (36 mo) |
| D-STOP50 | 54 | Dasatinib | MR4 | ≥1 yr | BCR-ABL1 > 0.0069% on 2 consecutive results | 57% (24 mo) |
| EURO-SKI39 | 755 | Imatinib > nilotinib/dasatinib | MR4 | ≥1 yr | Loss of MMR | 50% (24 mo) |
| Interim results | ||||||
| STIM2 (interim)53 | 124 | Imatinib | UMRD | ≥2 yr | Loss of MMR or ≥1-log increase in BCR-ABL1 | 59% (12 mo) |
| DASFREE (interim)54 | 84 | Dasatinib (first or second line) | MR4.5 | ≥1 yr | Loss of MMR | 49% (12 mo) |
| DESTINY55 | 117 | Imatinib > dasatinib/nilotinib | MR4 | ≥1 yr | Loss of MMR | 77% (24 mo) |
UMRD, undetectable molecular residual disease.
Subsequent stopping studies have had less stringent enrolment and restarting criteria. The A-STIM study, accepting loss of MMR as the trigger to TKI recommencement, showed a TFR rate of 61% at 36 months, establishing MMR as a safe threshold for TKI recommencement.42 The Euro-Ski study, the largest cessation study to date, allowed patients with MR4 for ≥1 year to cease TKI. At 24 months, 50% of patients attempting TFR remained in MMR off TKI.39 Discontinuation of second generation TKIs provided similar rates of success.38,43,44 Although the optimal depth and duration of DMR before TKI cessation are unclear, there is evidence to suggest that ≥24 months of sustained DMR is associated with superior TFR success.45
Molecular monitoring in TFR
Monitoring in the setting of DMR establishment and TFR should use high-quality standardized RT-qPCR46 (detection limit of at minimum MR4.5 if possible). The limit of detection of the method used needs to be clarified, because “PCR negativity” in a laboratory with a poor detection limit may lead to inappropriate TFR attempts. Cessation studies have predominantly incorporated monthly BCR-ABL1 testing for the first 6 to 12 months followed by 3-month testing thereafter. The timing of molecular relapse after TKI discontinuation is mainly within the first 6 months (Figure 1), reinforcing the requirement for the intensity of monitoring during this timeframe. Beyond 6 months, less frequent monitoring (ie, 2-3 months) could follow, because late molecular relapses are infrequent. However, monthly monitoring should ensue in the event of rising BCR-ABL1 transcripts. Loss of MR4.5 3 months after TKI cessation has been shown to predict TFR failure,44 although there is a small subset of patients who lose DMR but maintain MMR with fluctuating BCR-ABL1 transcript levels (Figure 1). Although late relapses are infrequent, there are reports of patients losing MMR and requiring TKI recommencement >36 months after drug cessation.44 This shows the importance of continued molecular monitoring every 3 to 6 months over the long term, despite apparent TFR success.
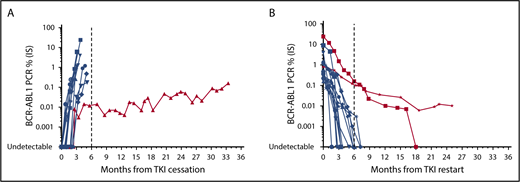
Molecular profile of 15 patients at our institution attempting and failing TFR. (A) Molecular relapse after TKI cessation. (B) Molecular response after TKI recommencement. Blue indicates typical molecular responses showing rapid rise or decline. Red indicates typical molecular responses showing slow rise or decline.
Molecular profile of 15 patients at our institution attempting and failing TFR. (A) Molecular relapse after TKI cessation. (B) Molecular response after TKI recommencement. Blue indicates typical molecular responses showing rapid rise or decline. Red indicates typical molecular responses showing slow rise or decline.
TKI recommencement in TFR failure
In the event of molecular relapse and MMR loss, we recommend 1- to 2-month BCR-ABL1 testing until MMR has been re-established. In nearly all cases, patients recommencing TKI after TFR failure remain sensitive to TKI therapy, with rare reports of KD mutation development.43,47 There has been a single report of progression to lymphoid blast crisis after TFR failure that occurred 9 months after imatinib recommencement, despite regaining MMR.42 However, the majority of patients rapidly regain MMR and DMR. Median times to both targets were 2 and 3 months, respectively, in the interim analysis of the STOP 2G-TKI study.44 Comparable data have been generated from other stopping studies,39,40,42,43,48,49 showing that, although most patients rapidly regain MMR and DMR, there remains a small subset of patients who fail to do so (Figure 1). Failure to regain MMR within 6 to 12 months of treatment initiation should prompt KD mutation analysis, and patients could appropriately be considered for a switch in TKI therapy.
Predictors of TFR success
Duration of TKI exposure before TFR has been linked to TFR success. Multivariate analysis of the STIM study revealed that imatinib treatment of >54 months was associated with lower risk of detectable BCR-ABL1 transcripts after TKI cessation,40 with similar results observed in other clinical trials.39,47,49 History of imatinib resistance in patients who have attempted TFR while on second generation TKIs has also been associated with an increased rate of molecular relapse.38,44 Prognostic models, such as the Sokal risk score, developed in the pre-TKI era remain useful as an indicator of the underlying biological characteristics at diagnosis and have been used to predict TFR success; low Sokal risk is associated with reduced likelihood of molecular relapse compared with those deemed high risk,40,41 but this has not been observed in all studies.39,50
Longer duration of DMR has been correlated with TFR success in both the Euro-Ski (>3.1 years39 ) study and the KID study (>36 months49 ), although this has not been replicated in other cessation trials.43,50 Furthermore, in the Euro-Ski study, each additional year of DMR was associated with a 3% increase in the probability of remaining in MMR at 6 months.39 Stability of DMR before TKI cessation may be an important variable in TFR success, but limited published data exist to date.42,51 Although no individual variable has been consistently predictive of TFR success, they all need to be considered before TFR attempts, and patients need to be counseled about their risk of molecular relapse before ceasing TKI.
Pitfalls associated with TFR
Careful patient selection is vital in maximizing TFR success but also, minimizing risk of adverse outcomes. Inappropriate cessation in patients with atypical transcripts can result in failure to detect molecular relapse. Inadvertent monitoring of the incorrect transcript can delay detection of relapse until hematological relapse.47 TFR attempts require reliable laboratory facilities able to provide rapid BCR-ABL1 results to ensure that clinicians can promptly intervene in cases of molecular relapse. Patient compliance remains vital, because adherence to the proposed monitoring schedule will minimize delayed interventions. Systems should exist to identify patients failing to comply with the monitoring regime and ensure timely clinician review of each BCR-ABL1 result.46
Pregnancy
Patients and their families need to be counseled against conception while on TKI therapy, and fertility preservation should be discussed with those of child-bearing age before TKI commencement. Because of the risk of birth deformities and miscarriage, TKIs should be ceased before conception and are not recommenced until after delivery. The timing of pregnancy should ideally be when the patient is eligible for a TFR attempt (ie, reached and maintained DMR for at least 12 months), but delaying TKI cessation to achieve this may not always be possible. The risk of disease progression and the importance of molecular monitoring during pregnancy need to be extensively discussed. We recommend monthly monitoring in pregnant patients, and after the BCR-ABL1 value approaches 1%, alfa interferon can be used until delivery has occurred. TKI can be promptly recommenced after delivery as long as breastfeeding is avoided.
Case discussion
Case 1
It was decided to persist with nilotinib rather than switch therapy. This was based on the significant fall in BCR-ABL level over the first 3 months and the fact that he was already on a potent TKI. Given identical molecular results in the context of imatinib therapy, we would have elected to switch to a second generation TKI.
This patient continued on nilotinib 300 mg twice daily, and at 6 months, his BCR-ABL1 value was 14% (IS). Having failed nilotinib therapy, the options were to switch to another second generation TKI, switch to ponatinib, or proceed with an allogeneic transplant. Based on the low rate of response to a second generation TKI after failure of another second generation TKI,52 ponatinib was the preferred choice. However, he was not eligible for ponatinib under the local funding scheme, and therefore, he was switched to dasatinib. He had minimal response to dasatinib, and within 3 months, his BCR-ABL1 increased to 53% (IS). Mutation analysis revealed that the pan-TKI–resistant T315I mutation had developed, and the patient was switched to ponatinib with an allograft planned. Ponatinib failed to achieve a better response, and 3 months later, the patient proceeded to a sibling-matched allogeneic stem cell transplant, after which his BCR-ABL1 transcripts became undetectable. This patient is a rare example of primary resistance to all TKI therapy in CML, and regular monitoring was key in identifying failure of milestone achievement and mutation development and vital in early recognition that allogeneic transplantation was the only option for this patient.
Case 2
This patient, although having achieved MMR by 18 months, would like to achieve TFR before contemplating pregnancy. Data presented above show that this patient is unlikely to achieve DMR on imatinib within a reasonable time.15,28 Because safe drug cessation is paramount and her timeframe is relatively short, either nilotinib or dasatinib would be a better option for her, with current evidence showing TFR success with second-line second generation TKI.43,44,47 This patient was switched to nilotinib and rapidly achieved MR4.5. She was able to attempt TFR within 2.5 years and has remained off therapy for over 12 months.
Case 3
Although the majority of patients regain MMR and DMR rapidly, there remains a small subset who have delayed response to TKI recommencement, which this case illustrates. The approach here could be to either persist with the current TKI after ensuring that no KD mutation has developed or switch to a different TKI, especially if a second TFR attempt is being considered. Given his initial very slow decline in BCR-ABL after restarting TKI, it was decided to switch to a more potent agent. This patient had no evidence of KD mutations and was switched to dasatinib 100 mg daily. He achieved MMR and MR4.5 within 3 months and has remained in MR4.5 for >2 years.
Conclusion
Because of the abundance of therapies available for patients with CML and the excellent outcomes achieved in most cases, there is a danger that CML may be regarded as an easy disease to treat, even by the nonexpert. This attitude can leave the high-risk and complex patient vulnerable to inadequate attention to toxicity issues, delayed therapeutic responses resulting in nonoptimal management, and inferior outcomes. To ensure that outcomes are optimized, regular high-quality molecular monitoring is absolutely vital in maximizing the number of patients who meet milestones that minimize the risk of progression and maximize the opportunity for TFR. Early detection of rising BCR-ABL1 values or failure to achieve predetermined milestones should trigger a thorough evaluation regarding compliance. KD mutation analysis may be required, and if patients fail therapy, a switch to a more potent TKI is required. The current awareness of TFR as a safe and feasible option has resulted in a shift in therapeutic goals for many patients, introducing the potential for an “operational cure” for CML. Achievement of sustained DMR correlates with TFR success, and maximizing DMR through appropriate therapy selection has become an increasingly important therapeutic target.
Acknowledgments
The authors thank Susan Branford and Verity Saunders for reviewing the manuscript.
N.S. receives scholarship funding from the Royal Adelaide Hospital Research Foundation Dawes Scholarship. T.P.H. is a Principal Research Fellow of the Australian National Health and Medical Research Council. This work was undertaken with the financial support of the South Australian Cancer Council’s Beat Cancer Project on behalf of its donors and the State Government through the Department of Health.
N.S. is a PhD candidate at the University of South Australia, and this work is submitted as partial fulfilment of the university requirements.
Authorship
Contribution: N.S. and T.P.H. wrote and reviewed the manuscript.
Conflict-of-interest disclosure: N.S. has received honoraria from Novartis, Bristol-Myers Squibb, and Janssen. T.P.H. has received research funding and honoraria and consulted for Novartis, Bristol-Myers Squibb, and Ariad. Off-label drug use: None disclosed.
Correspondence: Naranie Shanmuganathan, Department of Haematology, Royal Adelaide Hospital, Port Rd, Adelaide, SA 5000, Australia; e-mail: naranie.shanmuganathan@sa.gov.au.
