

TO THE EDITOR:
Acute lymphoblastic leukemia (ALL) is the most common pediatric malignancy, and T-cell ALL (T-ALL) accounts for approximately 15% of cases of childhood ALL.1,2 Early T-cell precursor ALL (ETP-ALL) is a recently recognized subtype of T-ALL.3,4 Hallmarks of T-ALL include genetic alterations of T-cell transcription factors and components involved in the Notch signaling pathway and the JAK-STAT pathway as well as epigenetic regulators.5,6 SUZ12, EED, and EZH2 are the core components of polycomb repressive complex 2 (PRC2), which catalyzes the trimethylation of H3 at lysine 27 (H3K27me3). PRC2 is a critical regulator of normal hematopoiesis, and mutations in PRC2 have been identified in many hematologic malignancies.7 Loss-of-function mutations of EZH2 and SUZ12 are found in 25% of T-ALL cases, whereas 42% of ETP-ALL cases harbor deletions or sequence mutations of SUZ12, EED, and EZH2.4 In mouse models, EZH2 deficiency was reported to induce T-ALL and activate the JAK-STAT signaling pathway.8-10 In addition, EZH2 deletion induced activation of STAT3 in a murine ETP-ALL model, and ruxolitinib, an inhibitor of the JAK-STAT signaling pathway, could be used to treat ETP-ALL.11
In our study, targeted sequencing was used to detect mutations of the PRC2 core components (EZH2, SUZ12, and EED) in 60 primary T-ALL samples from the Pediatrics Department of the Blood Diseases Hospital (Chinese Academy of Medical Sciences), and 14 mutations in EZH2 (8 of 60), SUZ12 (4 of 60), and EED (2 of 60) were discovered in 12 patients (Figure 1A). Neither copy number variations of EZH2, SUZ12, and EED (detected by droplet digital polymerase chain reaction [PCR]) nor FLT3 pathway component mutations or variations were observed in these 12 patients (data not shown). Retrospective analysis revealed that T-ALL patients with PRC2 mutations were inclined to have higher numbers of bone marrow blasts than those without PRC2 mutations (median, 88.5% vs 81.72%; P = .026). Consistent with a previous study,4 these mutations were detected in 42% (5 of 12) of patients with ETP-ALL in contrast to only 15% (7 of 48) of patients who did not have ETP-ALL (supplemental Table 1, available on the Blood Web site).
![Figure 1. Decreased H3K27me3 enrichment caused by PRC2 disruption strikingly enhances the activation of FLT3 and therefore the downstream pathway. (A) Hierarchical clustering profile of the top 60 differentially expressed genes (P < .001) by whole-transcriptome sequencing. Columns indicate genes, and rows represent PRC2Mut patients. The normalized expression level for each gene (z-score) is indicated by a color (red for overexpression and blue for underexpression). PRC2 genetic makeup and T-ALL subtype of each patient are also shown. (B-D) FLT3 messenger RNA level verified in T-ALL, ETP-ALL, and non-ETP-ALL patients. (E) FLT3 expression of leukemic cells from 4 T-ALL patients: TJCT003 (EZH2Mut-P1), TJCT006 (EZH2Mut-P2), TJCT021 (EZH2WT-P2 [PRC2 WT]), and TJCT037 (EZH2WT-P1 [PRC2 WT]) detected by flow cytometry. (F-G) FLT3 expression and phosphorylation in EZH2-knockout (EZH2-KO) (Δ/Δ and WT/Δ) and EZH2WT Jurkat cell clones. (H) EZH2 expression, H3K27me3 modification, and FLT3 downstream signaling pathway analyzed by western blotting in EZH2-KO (Δ/Δ and WT/Δ) and EZH2WT Jurkat cell clones. (I-J) H3K27me3 (I) mark and (J) POLII enrichment at the transcriptional start site of FLT3 measured by chromatin immunoprecipitation quantitative PCR in EZH2 KO cells (JE043 and JE010 clones). Normal rabbit immunoglobulin G (IgG) was used as the control. (K) FLT3 expression levels and downstream pathway activities assessed in EZH2 WT (Jurkat-Cas9), heterozygous KO (JE043), and homozygous KO (JE010) Jurkat cells after being treated with vehicle (dimethyl sulfoxide [DMSO]), sorafenib (10 nM), and quizartinib (10 nM) for 24 hours. All the WT and EZH2 KO Jurkat cells were cultured with normal medium (RPMI-1640 and 10% fetal bovine serum [FBS]) and supplied with FLT3 ligand (5 ng/mL). All experiments were performed in triplicate. Data are presented as the mean ± standard deviation (SD). Error bars: SD of 3 independent experiments. ***P < .001. ns, not significant.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/132/23/10.1182_blood-2018-04-845628/6/m_blood845628f1.png?Expires=1769121011&Signature=yU4~iMU4k7mMf0NEqvrPaMyv5iF3-XCOkD5pD80I~CkHFHMJ3-ZxHoExSHvjDBlEoDlEy00ggI4hWP8sGBRmDKmOtIORAo9hp-cGNpq1yvYeX0dk88GJF59UsHZPsl-2~nna8rb81fSmPaUhU-2FvqzszuKIEyIhRdKNm1~vp8Te0TubTX5IVcs9jCcoA1lMCFl71JqxkqKsXhr~KFJANuywkv-wc2kLFzENH~ENDwp5aHk3txJA7kSO7fzjjTFDLIhl24pX9HN~QP7sq9HvdTk7G7QkcEBIijbRkLCVtJF3j6S2h5sdv3omIVrXCMJR4MhdGRBzMZpka0ud8Jtjbg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Decreased H3K27me3 enrichment caused by PRC2 disruption strikingly enhances the activation of FLT3 and therefore the downstream pathway. (A) Hierarchical clustering profile of the top 60 differentially expressed genes (P < .001) by whole-transcriptome sequencing. Columns indicate genes, and rows represent PRC2Mut patients. The normalized expression level for each gene (z-score) is indicated by a color (red for overexpression and blue for underexpression). PRC2 genetic makeup and T-ALL subtype of each patient are also shown. (B-D) FLT3 messenger RNA level verified in T-ALL, ETP-ALL, and non-ETP-ALL patients. (E) FLT3 expression of leukemic cells from 4 T-ALL patients: TJCT003 (EZH2Mut-P1), TJCT006 (EZH2Mut-P2), TJCT021 (EZH2WT-P2 [PRC2 WT]), and TJCT037 (EZH2WT-P1 [PRC2 WT]) detected by flow cytometry. (F-G) FLT3 expression and phosphorylation in EZH2-knockout (EZH2-KO) (Δ/Δ and WT/Δ) and EZH2WT Jurkat cell clones. (H) EZH2 expression, H3K27me3 modification, and FLT3 downstream signaling pathway analyzed by western blotting in EZH2-KO (Δ/Δ and WT/Δ) and EZH2WT Jurkat cell clones. (I-J) H3K27me3 (I) mark and (J) POLII enrichment at the transcriptional start site of FLT3 measured by chromatin immunoprecipitation quantitative PCR in EZH2 KO cells (JE043 and JE010 clones). Normal rabbit immunoglobulin G (IgG) was used as the control. (K) FLT3 expression levels and downstream pathway activities assessed in EZH2 WT (Jurkat-Cas9), heterozygous KO (JE043), and homozygous KO (JE010) Jurkat cells after being treated with vehicle (dimethyl sulfoxide [DMSO]), sorafenib (10 nM), and quizartinib (10 nM) for 24 hours. All the WT and EZH2 KO Jurkat cells were cultured with normal medium (RPMI-1640 and 10% fetal bovine serum [FBS]) and supplied with FLT3 ligand (5 ng/mL). All experiments were performed in triplicate. Data are presented as the mean ± standard deviation (SD). Error bars: SD of 3 independent experiments. ***P < .001. ns, not significant.
Decreased H3K27me3 enrichment caused by PRC2 disruption strikingly enhances the activation of FLT3 and therefore the downstream pathway. (A) Hierarchical clustering profile of the top 60 differentially expressed genes (P < .001) by whole-transcriptome sequencing. Columns indicate genes, and rows represent PRC2Mut patients. The normalized expression level for each gene (z-score) is indicated by a color (red for overexpression and blue for underexpression). PRC2 genetic makeup and T-ALL subtype of each patient are also shown. (B-D) FLT3 messenger RNA level verified in T-ALL, ETP-ALL, and non-ETP-ALL patients. (E) FLT3 expression of leukemic cells from 4 T-ALL patients: TJCT003 (EZH2Mut-P1), TJCT006 (EZH2Mut-P2), TJCT021 (EZH2WT-P2 [PRC2 WT]), and TJCT037 (EZH2WT-P1 [PRC2 WT]) detected by flow cytometry. (F-G) FLT3 expression and phosphorylation in EZH2-knockout (EZH2-KO) (Δ/Δ and WT/Δ) and EZH2WT Jurkat cell clones. (H) EZH2 expression, H3K27me3 modification, and FLT3 downstream signaling pathway analyzed by western blotting in EZH2-KO (Δ/Δ and WT/Δ) and EZH2WT Jurkat cell clones. (I-J) H3K27me3 (I) mark and (J) POLII enrichment at the transcriptional start site of FLT3 measured by chromatin immunoprecipitation quantitative PCR in EZH2 KO cells (JE043 and JE010 clones). Normal rabbit immunoglobulin G (IgG) was used as the control. (K) FLT3 expression levels and downstream pathway activities assessed in EZH2 WT (Jurkat-Cas9), heterozygous KO (JE043), and homozygous KO (JE010) Jurkat cells after being treated with vehicle (dimethyl sulfoxide [DMSO]), sorafenib (10 nM), and quizartinib (10 nM) for 24 hours. All the WT and EZH2 KO Jurkat cells were cultured with normal medium (RPMI-1640 and 10% fetal bovine serum [FBS]) and supplied with FLT3 ligand (5 ng/mL). All experiments were performed in triplicate. Data are presented as the mean ± standard deviation (SD). Error bars: SD of 3 independent experiments. ***P < .001. ns, not significant.
RNA sequence profiling of the leukemia cells from the 12 patients with PRC2 mutations revealed significant upregulation of FLT3 (Figure 1A), and this upregulation was corroborated to present specifically in PRC2-mutated (PRC2Mut) patients by analysis with quantitative reverse transcription PCR (qRT-PCR) analysis, irrespective of ETP or non-ETP-ALL in this group (Figure 1B-D). The expression of FLT3 in EZH2-mutant (EZH2Mut) patients was slightly higher than that in SUZ12 or EED patients but not significantly higher (supplemental Figure 1). Xenografts were established in NOD/SCID mice to initiate patient-derived leukemia cells, which were isolated from 2 EZH2Mut patients (P1 and P2) and 2 non-PRC2Mut (EZH2 wild-type [EZH2WT]; P1 and P2) T-ALL patients. The expressions of EZH2 were confirmed to be similar within these 4 patients (supplemental Figure 2). Nonetheless, FLT3 was significantly highly expressed in EZH2Mut patients (Figure 1E; supplemental Figure 3). Next, to explore the mechanism of aberrant expression of FLT3 in patients with PRC2Mut T-ALL, we knocked out EZH2 in the Jurkat cell line via CRISPR/Cas9. Three heterozygous and 3 homozygous EZH2 knockout clones were chosen for analysis (supplemental Figure 4). Western blot verified that deletions of EZH2 abrogated the H3K27me3 modification, accentuated expression and phosphorylation of FLT3, and elevated the activity of STAT5, AKT, and ERK signaling pathway in EZH2 knockout Jurkat cells (Figure 1F-H; supplemental Figure 5). Chromatin immunoprecipitation quantitative PCR analysis of the homozygously deleted cell lines showed that EZH2 deletions led to strongly attenuated H3K27me3 and prominently enriched POLII binding at the transcription start site of the FLT3 gene, consistent with the gene expression levels (Figure 1I-J). To further elucidate the regulation of FLT3 by EZH2 in T-ALL cells, we knocked down EZH2 in Jurkat, CEM, and Molt4 cells, and by so doing we also observed the upregulation of FLT3, phosphorylation of FLT3, and activated downstream STAT5, AKT, and ERK signaling pathways (supplemental Figure 6). We overexpressed EZH2 in a well-reported EZH2 low-expression cell line (Loucy12 ) and demonstrated that EZH2 overexpression in Loucy cells attenuated the expression of FLT3 as well as the downstream signaling pathway (supplemental Figure 7). In addition, we found that overexpression of FLT3 in Jurkat cells activated the downstream signaling pathway, which coincided with EZH2 deletions in Jurkat cells (supplemental Figure 8). These results further verified the regulation of FLT3 and downstream signaling pathways by EZH2 in T-ALL cells. Of note, we observed that elevated phosphorylation of STAT5, AKT, and ERK in EZH2 knockout cells was reversed by treatment with FLT3 inhibitors (sorafenib and quizartinib) (Figure 1K; supplemental Figure 9). Regarding the heterozygous clone, 10 nM quizartinib shifted the hyperactivated phosphorylation back toward a nearly normal level, which raised the notion that the heterozygous mutants hinted of being a potential target for FLT3 inhibitors, although future clinical observation remains open. We next exposed cell lines of T-lineage (Jurkat), B-lineage (Ramos), and myeloid malignancies (K562, HL-60, THP-1) to EZH2 inhibitors (EPZ005687 and GSK343) and observed that only T-lineage cell lines responded to EZH2 inhibitors with increased expression of FLT3 and activation of the downstream pathway (supplemental Figure 10). An in vitro T-lineage differentiation assay was used to investigate the roles that FLT3 and PRC2 played in T-cell development. In this assay, immature T-cell differentiation was induced from murine lineage–Sca1+c-Kit+ (LSK) bone marrow cells cocultured with OP9-DL1 cells (supplemental Figure 11). The results showed that EZH2 inhibition led to the arrest of marked T-lineage differentiation and upregulation of the FLT3 downstream pathway.
The 4 established patient-derived xenograft (PDX) mouse models (Figure 1C) were used to determine whether aberration in the FLT3 downstream pathway was a potential target for patients with PRC2Mut T-ALL. Whole-exome sequencing proved no FLT3 or FLT3 signaling–related cooperative mutations in these 4 patients, where the xenografts originated (supplemental Table 2). Surprisingly, pharmacologically achievable concentrations of sorafenib and quizartinib strikingly prolonged the survival of EZH2Mut, but not EZH2WT, T-ALL recipient mice (Figure 2A-D). By cytometric analysis with primary CD45+ cells isolated from PDX mice after 5 days of treatment with sorafenib or quizartinib was completed, we observed that FLT3 inhibitors specifically exerted attenuation on the FLT3 downstream signaling pathway in leukemic cells from EZH2Mut-engrafted mice but barely affected the stability of phosphorylated pSTAT5, pAKT, or pERK in EZH2WT leukemic cells (Figure 2E; supplemental Figure 12).
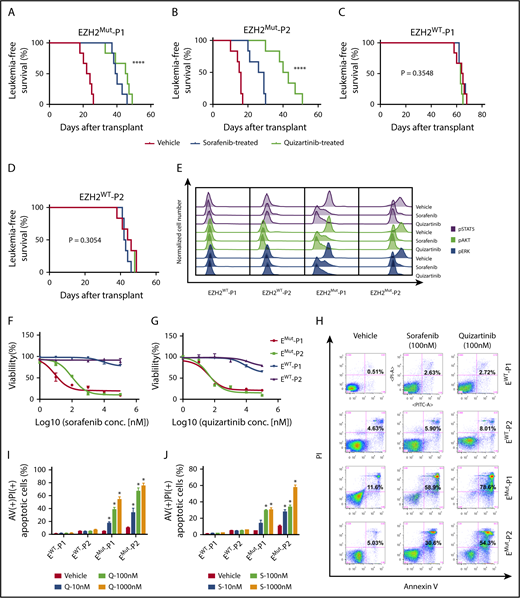
Administration of FLT3 inhibitors effectively induces apoptosis of EZH2Mut leukemic cells in vitro and prolongs the lifespan of a xenograft mouse model with anti-leukemia activity in vivo. (A-D) Kaplan-Meier plots of survival in NOD-SCID mice inoculated with (A-B) EZH2Mut or (C-D) EZH2WT T-ALL patient-derived leukemic cells via the tail vein. When the leukemic proportion extended up to 5% in peripheral blood, mice were intraperitoneally injected with vehicle (DMSO), sorafenib (60 mg/kg), or quizartinib (30 mg/kg) for 5 consecutive days. Each treatment group contained 5 animals. Day 0 was the day when cells were implanted. (E) Flow cytometric analyses of phosphorylated pSTAT5, pAKT, and pERK performed for isolated tumor cells from patient-derived xenograft mice (EZH2Mut and EZH2WT) after being treated with vehicle (DMSO), sorafenib, or quizartinib for 5 days. (F-G) Cell growth and viability of tumor cells (harvested from drug-free xenograft mice) assessed in vitro by MTT assays after being treated with designated concentrations of (F) sorafenib or (G) quizartinib for 24 hours in parallel studies. (H-J) Cell death analyzed in the bulk blast population using an annexin V (AV)/propidium iodide (PI) staining assay in vitro after being exposed to sorafenib or quizartinib for 24 hours. Additional analyses of the late-stage apoptotic cell percentage were carried out by concentration titrated (I) sorafenib and (J) quizartinib. All the cells harvested for in vitro studies were incubated in RPMI-1640 with 10% FBS and 5 ng/mL FLT3 ligand overnight (16 hours) for synchronization before treatment started. Data are presented as the mean ± SD. Error bars: SD of 3 independent experiments. *P < .05; ****P < .00001.
Administration of FLT3 inhibitors effectively induces apoptosis of EZH2Mut leukemic cells in vitro and prolongs the lifespan of a xenograft mouse model with anti-leukemia activity in vivo. (A-D) Kaplan-Meier plots of survival in NOD-SCID mice inoculated with (A-B) EZH2Mut or (C-D) EZH2WT T-ALL patient-derived leukemic cells via the tail vein. When the leukemic proportion extended up to 5% in peripheral blood, mice were intraperitoneally injected with vehicle (DMSO), sorafenib (60 mg/kg), or quizartinib (30 mg/kg) for 5 consecutive days. Each treatment group contained 5 animals. Day 0 was the day when cells were implanted. (E) Flow cytometric analyses of phosphorylated pSTAT5, pAKT, and pERK performed for isolated tumor cells from patient-derived xenograft mice (EZH2Mut and EZH2WT) after being treated with vehicle (DMSO), sorafenib, or quizartinib for 5 days. (F-G) Cell growth and viability of tumor cells (harvested from drug-free xenograft mice) assessed in vitro by MTT assays after being treated with designated concentrations of (F) sorafenib or (G) quizartinib for 24 hours in parallel studies. (H-J) Cell death analyzed in the bulk blast population using an annexin V (AV)/propidium iodide (PI) staining assay in vitro after being exposed to sorafenib or quizartinib for 24 hours. Additional analyses of the late-stage apoptotic cell percentage were carried out by concentration titrated (I) sorafenib and (J) quizartinib. All the cells harvested for in vitro studies were incubated in RPMI-1640 with 10% FBS and 5 ng/mL FLT3 ligand overnight (16 hours) for synchronization before treatment started. Data are presented as the mean ± SD. Error bars: SD of 3 independent experiments. *P < .05; ****P < .00001.
In vitro MTT assays from primary CD45+ cells manifested EZH2Mut but not EZH2WT leukemic cells, which had conferred sensitivity to sorafenib or quizartinib treatment (Figure 2F-G). Further investigation revealed that administration of sorafenib or quizartinib in vitro dramatically induced tumor cell apoptosis and suppressed the excess phosphorylation of STAT5, AKT, and ERK in EZH2Mut T-ALL cells (Figure 2H-J; supplemental Figure 13). These results demonstrated that hyperactive FLT3 expressions as well as the downstream signaling pathway were essential for maintenance of PRC2Mut T-ALL cells.
A previous study demonstrated that FLT3 inhibitors induced apoptosis in ∼30% of cells from patients with FLT3 high-expression T-ALL, and synergistic inhibition of FLT3 and KIT effectively suppressed the other 70% of cells from patients with FLT3 high-expression T-ALL.13 However, FLT3 has not been shown to contribute to leukemogenesis in PRC2-inactivated T-ALL. EZH2 deletions have been highlighted in previous studies as a potential target of JAK-STAT pathway inhibition in murine ETP-ALL.8 Our study identified and characterized another novel oncogenic mechanism involving increases in FLT3 transcription and the downstream signaling pathway resulting from the loss of H3K27me3 from PRC2 inactivation in T-ALL. Significantly, our in vivo study further demonstrates that the aberrant FLT3 downstream pathway is a susceptible therapeutic target for PRC2-inactivated T-ALL, which provides new insights into precision therapy for T-ALL.
The online version of this article contains a data supplement.
Acknowledgments
The authors thank the patients for participating in this study.
This work was supported by Ministry of Science and Technology of China grant 2016YFA0100600 (T.C.); Chinese Academy of Medical Sciences Innovation Fund for Medical Sciences grants 2016-I2M-1-002 (X.Z.), 2017-I2M-1-015 (Y. Zhang), 2016-I2M-1-017 (T.C.), 2017-I2M-3-018 (Y. Chen), and 2017-I2M-3-009 (P.W.); National Nature Science Foundation of China grants 81421002 (T.C. and X.Z.), 81470339 (X.Z.), 81400137 and 81770175 (Y. Zhang), 81672010 (Y. Cao), and 31600705 (J.Y.); the Peking Union Medical College Youth Fund grant 3332016091 (Y. Zhang); and the nonprofit Central Research Institute Fund of the Chinese Academy of Medical Sciences grant 2016ZX310184-1 (Y. Zhang).
Authorship
Contribution: Y. Zhang, X.Z., T.C., and Y. Cao designed the research; J.Z., M.Z., X.L., J.Y., and C.L. performed the experiments; X.C., W.Y., L.Z., Y.G., Y. Zou, and Y. Chen recruited patients and performed follow-up; P.W. performed the bioinformatic analysis and interpretation; J.Z., Y. Zhang, J.Y., and M.Z. analyzed the results; and Y. Zhang, X.Z., Y. Cao, J.Y., and T.C. wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Yingchi Zhang, Chinese Academy of Medical Sciences and Peking Union Medical College, 288 Nanjing Rd, Tianjin 300020, China; e-mail: zhangyingchi@ihcams.ac.cn; Tao Cheng, Chinese Academy of Medical Sciences and Peking Union Medical College, 288 Nanjing Rd, Tianjin 300020, China; e-mail: chengtao@ihcams.ac.cn; and Xiaofan Zhu, Chinese Academy of Medical Sciences and Peking Union Medical College, 288 Nanjing Rd, Tianjin 300020, China; e-mail: xfzhu@ihcams.ac.cn.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal