

In this issue of Blood, report a new method of comparing published murine studies assessing the impact of single gene defects on arterial thrombus formation, thromboembolism, and tail bleeding time. This approach is meant to obtain reliable information on the role of specific genes in thrombosis and hemorrhage in comparison with human orthologs.1
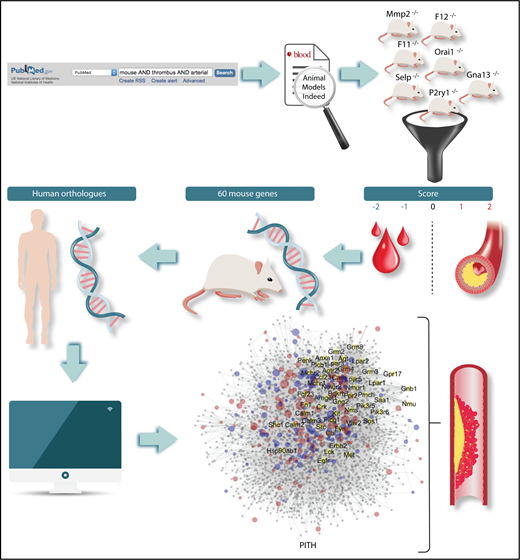
Flowchart of the methods for the identification of mouse genes relevant to thrombosis and hemostasis. The definition of human orthologs and the construction of a network of protein interactions in thrombosis and hemostasis (PITH) have then led to the detection of novel proteins with potential contribution to thrombosis and hemostasis.
Flowchart of the methods for the identification of mouse genes relevant to thrombosis and hemostasis. The definition of human orthologs and the construction of a network of protein interactions in thrombosis and hemostasis (PITH) have then led to the detection of novel proteins with potential contribution to thrombosis and hemostasis.
Atherothrombotic ischemic cardiovascular disease (CVD) remains the leading cause of death and disability worldwide and, despite great advances in prevention and treatment, continues to represent a heavy burden on public health. Therefore, research on the basic pathogenic mechanisms of arterial thrombosis is crucial for the development of more effective therapeutic strategies. The major shortcoming of the advancements made so far in antiplatelet treatments has been the increased hemorrhagic risk inseparably associated with increased antithrombotic efficacy.2-4 Thus, the search for novel targets preventing arterial thrombosis without altering hemostasis has generated much interest.5-8 The development of animal models of atherothrombotic CVD together with the progress of techniques for sophisticated genetic manipulation of mice has provided important tools for better understanding the pathophysiology of CVD, its genetic basis, and potentially for the identification of novel pharmacologic targets.
However, studies in genetically modified mice have several limitations that may undermine their extrapolation to human disease. These studies are typically carried out using small numbers of mice, applying disparate methodologies to assess thrombus formation and employing different measurement outcomes, rendering the results difficult to assess and compare and to extrapolate to humans. Moreover, several limitations inherent to mouse studies must be considered. Inbred mouse strains are genetically homogeneous in contrast to the wide genetic heterogeneity of the human population. These mice are carefully protected from the exposure to microorganisms and thus have a “standardized” gut microbiome, differently from humans, who have a widely varied gut microbiome, which may strongly influence the thrombotic risk.9 Mice also lack comorbidities that are typical of patients with CVD.10
To overcome these limitations, Baaten and coworkers have carried out an encyclopedic analytical examination of the literature, assessing 1431 mouse thrombosis studies published over the last 3 decades dealing with 409 mouse genes. They have developed an original scoring procedure according to which studies were grouped by end point (arterial thrombosis, thromboembolism, or tail bleeding time), and the observed alterations were graded on a 3-point or 5-point scale. Scores were then grouped per gene, providing a list of 60 mouse genes for which there was consistent evidence for a role in (1) arterial thrombosis but not hemostasis, (2) both thrombosis and hemostasis, and (3) thrombosis accompanied with prolonged bleeding (see figure). The human orthologs of 35 of these genes were known to be involved in human disease, but for an additional 25 genes, the relationship to human diseases has not been established. Interestingly, this approach revealed 17 candidate proteins with a possible role in arterial thrombosis without an effect on bleeding. Although for some of these a role in thrombosis was expected (eg, FXI and FXII, for which antagonists are already being tested as potential antithrombotic agents), other unforeseen targets have emerged, like the P2X purinergic receptor, the GTPase RhoG, the member of extracellular matrix adhesion receptors integrin α6, and the regulator of platelet dense granule biogenesis SGK1.
The authors also built a network of protein interactions in thrombosis and hemostasis, based on the mouse genes found to be implicated in thrombus formation or bleeding. In this way, the authors identified additional genes expressed by megakaryocytes of potential pathophysiologic relevance (see figure). The authors then tested experimentally the role of some of these proteins in thrombus formation using knockout mice and revealed that 4 of them (Apoe, Fpr2, Ifnar1, Vps13a) play a significant role. Indeed, knockout mice for Ifnair1 and Vps13a show decreased arterial thrombus formation, whereas mice lacking ApoE and Frp2 show increased thrombus formation. The results of this study have multiple implications: they show that, despite the important limitations, studies in genetically modified mice provide reasonably consistent results; they identify unexpected pathways that may play a role in thrombosis and hemostasis; they unravel new targets of potential interest for antithrombotic therapy; they show that the ex vivo measurement of thrombus formation using flow chambers mimics rather faithfully thrombus formation in vivo, suggesting that this method, after appropriate standardization and automatization, may become an excellent tool for antithrombotic drug testing.
The study by Baaten and coworkers represents the first significant attempt to quantitatively assess gene effects on thrombosis and hemostasis in mice. This study provides important insight into the strengths and weaknesses of mouse models for thrombosis research and will probably become a precious source of information on gene-modified mouse models in the field.
Conflict-of-interest disclosure: The authors declare no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal