

Key Points
A synthesis method was developed comparing the roles of 431 mouse genes in arterial thrombosis with or without affecting hemostasis.
A network of protein interactions in thrombosis/hemostasis revealed high similarity in mouse and man, and identified candidate proteins.

Abstract
Antithrombotic therapies reduce cardiovascular diseases by preventing arterial thrombosis and thromboembolism, but at expense of increased bleeding risks. Arterial thrombosis studies using genetically modified mice have been invaluable for identification of new molecular targets. Because of low sample sizes and heterogeneity in approaches or methodologies, a formal meta-analysis to compare studies of mice with single-gene defects encountered major limitations. To overcome these, we developed a novel synthesis approach to quantitatively scale 1514 published studies of arterial thrombus formation (in vivo and in vitro), thromboembolism, and tail-bleeding of genetically modified mice. Using a newly defined consistency parameter (CP), indicating the strength of published data, comparisons were made of 431 mouse genes, of which 17 consistently contributed to thrombus formation without affecting hemostasis. Ranking analysis indicated high correlations between collagen-dependent thrombosis models in vivo (FeCl3 injury or ligation/compression) and in vitro. Integration of scores and CP values resulted in a network of protein interactions in thrombosis and hemostasis (PITH), which was combined with databases of genetically linked human bleeding and thrombotic disorders. The network contained 2946 nodes linked to modifying genes of thrombus formation, mostly with expression in megakaryocytes. Reactome pathway analysis and network characteristics revealed multiple novel genes with potential contribution to thrombosis/hemostasis. Studies with additional knockout mice revealed that 4 of 8 (Apoe, Fpr2, Ifnar1, Vps13a) new genes were modifying in thrombus formation. The PITH network further: (i) revealed a high similarity of murine and human hemostatic and thrombotic processes and (ii) identified multiple new candidate proteins regulating these processes.
Introduction
Cardiovascular disease, due to vaso-occlusive arterial thrombosis or thromboembolism, is a leading cause of disability and death worldwide.1 Extensive research over the last decades has provided drugs that result in a significant, albeit incomplete, reduction of disease-related mortality and morbidity. Apart from pharmacological agents lowering plasma lipids and blood pressure, antiplatelet and anticoagulant agents are effective in the secondary prevention of thrombotic complications.2-4 Yet, current treatment regimens are still only partly effective, while causing bleeding complications in at least 2% of the patients. This has led to a continued search for better targeted antithrombotic medication with minimal bleeding.
In the past 2 decades, hundreds of mouse studies have been performed to evaluate consequences of genetic modification or pharmacological intervention in experimental models of arterial thrombosis or thromboembolism.3,5,6 The connecting aim of such studies is to disclose particular platelet, coagulation, or vascular proteins that regulate thrombosis and/or hemostasis. However, these studies typically use low numbers of mice (typically 6-12), and assess arterial thrombus formation or thromboembolism in vivo or in vitro, using different methodologies and measurement outcomes. This variation has hampered a systematic comparison of the use of thrombosis models across genes. The few published reviews provide qualitative rather than quantitative descriptions of the effects of genetic modification on thrombus formation or tail-bleeding.3,5,7,8 On the other hand, many of the published effects of murine gene deletion faithfully phenocopy the known effects of Mendelian inherited bleeding and platelet disorders in humans, thus proving their relevance for disease.9,10 This prompted us to develop new means for quantitative comparison of published studies regarding altered thrombosis and bleeding tendencies as a consequence of genetic modification in mice.
Published reports on murine arterial thrombosis have in common an experimental approach, in which vascular damage is induced by activation or injury of the endothelium to cause exposure of the collagen-containing subendothelial matrix.10 Nevertheless, in vivo methods differ regarding (i) the type of injured vascular bed, (ii) the type and strength of the injury, (iii) equipment and detection methods, and (iv) outcome parameters. Hence, for systematic comparison of multiple studies, an analytical tool needs to be developed that is scaled and takes into account these variables. The same applies to in vitro tests, where microfluidics devices for whole blood perfusion are used as proxy measurements of arterial thrombus formation.11-13 Such in vitro tests are relevant from a translational point of view because the same devices can be used to detect deficiencies in human platelet and coagulation activity and to determine the efficacy of antithrombotic medication.13-17
In the present paper, we developed and validated a scaling procedure as a synthesis approach to compare published studies on effects of genetic modification on arterial thrombus formation, thromboembolism, and/or tail-bleeding. This synthesis gave an unprecedented insight into the relative roles of multiple mouse genes, in majority regulating platelet, coagulation, or vascular functions.
Methods
For a more extended description of the methods, see supplemental Methods.
A PubMed search over the years 1980 to 2018 was performed to identify papers on mouse studies in relation to arterial thrombus formation, thromboembolism, and tail-bleeding (supplemental Table 1). From these papers, information on genetics, experimental models, and outcome parameters was gathered (supplemental Data File 1).
As an alternative synthesis approach, 1514 studies (1431 studies published before January 2018) were grouped per experimental method, vascular bed of injury, and outcome thrombus parameter (mass-dependent, time-dependent and stability measurements, bleeding times) and then normalized on a 3-point or 5-point scale. Mean scores per group, parameter type, and gene were compared using the nonparametric Kendall τ-c test. To assess consistency and strength of scaled studies per gene, a consistency parameter (CP) was defined as 1 − log(P), with P values from a 1-sample Student t test.
Animals came from breeding programs of the Wellcome Sanger Institute Mouse Genetics Program (Cambridge, United Kingdom) and the Institute for Cardiovascular Prevention in Munich (Germany). Animal experiments were approved by the local animal experimental committees.
Results
Data collection of mouse studies reporting on genetic modification or pharmacological intervention affecting arterial thrombosis, thromboembolism, or tail-bleeding
A PubMed search was performed across scientific publications over the period 1980 to 2018, resulting in 610 publications containing quantified data (with statistics) on effects of genetic or pharmacological perturbation on arterial thrombus formation, thromboembolism, or tail-bleeding in mice. The majority of the papers concerned mice with a single genetic deficiency, resulting in altered platelet, coagulant, or vascular function. The published studies were classified (Figure 1) as in vivo arterial thrombosis (class I), in vivo thromboembolism (class II), in vitro arterial thrombus formation (class III), or tail-bleeding (class IV). Manuscripts as well as published reviews were screened for further primary sources, thereby including another 32 publications. Individual studies were defined as reporting on effects of genetic modification per vascular bed or way of injury, resulting in a database of 1514 studies (1431 studies published before January 2018) (supplemental Data File 1).
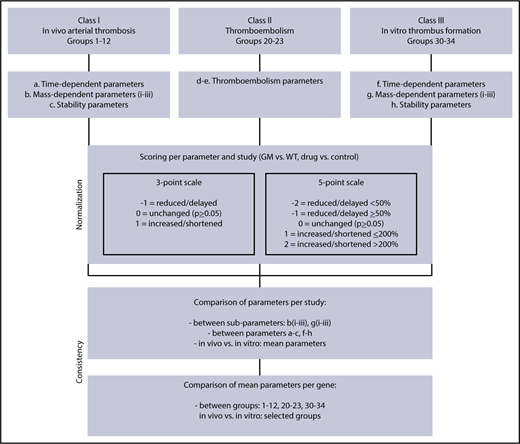
Flowchart for scaling of parameters of arterial thrombosis and thromboembolism. Scored were studies of in vivo arterial thrombus formation (class I), thromboembolism (class II), and in vitro thrombus formation (class III) for mice with a genetic modification (GM) or pharmacological treatment, in comparison with wild type (WT) or the control condition. Indicated are scaling rules (scores) for the various output parameters on 3- and 5-point scales (values for WT set at 100%). For detailed description of groups and scoring procedures, see supplemental Table 2.
Flowchart for scaling of parameters of arterial thrombosis and thromboembolism. Scored were studies of in vivo arterial thrombus formation (class I), thromboembolism (class II), and in vitro thrombus formation (class III) for mice with a genetic modification (GM) or pharmacological treatment, in comparison with wild type (WT) or the control condition. Indicated are scaling rules (scores) for the various output parameters on 3- and 5-point scales (values for WT set at 100%). For detailed description of groups and scoring procedures, see supplemental Table 2.
Conventional meta-analysis and limitations
As a first approach to quantitatively compare outcomes of thrombosis studies per mouse gene, a conventional meta-analysis was performed for the most studied mouse strains. Given the diversity between studies (thrombosis model, detection method, and parameter measured), a standard random-effects model was considered most appropriate. To achieve a certain degree of interstudy consistency, only studies were included where FeCl3 was used to induce thrombosis in either the carotid, mesenteric, or femoral arteries/arterioles, and where time-dependent parameters were reported (see supplemental Methods). For 6 genes, data could thus be obtained from a small number (n) of studies: Cd36 (4), Clec1b (3), F12 (4), Fcer1g (2), Gp6 (6), and Vwf (3) (Figure 2). Meta-analysis per gene, using the Cochrane group RevMan 5.1 program, pointed to a significantly prolonged thrombus formation in mice with genetic deficiency in F12, Gp6, or Vwf (P = .0008-.003), whereas there was a tendency to prolongation in mice lacking Cd36, Clec1b, or Fcer1g (P = .05-.08). A limitation of this meta-analysis approach, however, is the large heterogeneity index for all genes (I2 = 73%-88%), even when statistical significance is reached. We reasoned that this high heterogeneity is unavoidable because animal experiments are de facto quite small in size, and furthermore vary in experimental conditions and outcome parameters. We further noticed that a heterogeneity test is low in power, when sample size is small and few trials are included.18 This prompted us to search for a better method to compare and normalize effects of genetic knockout in a larger set of published studies.
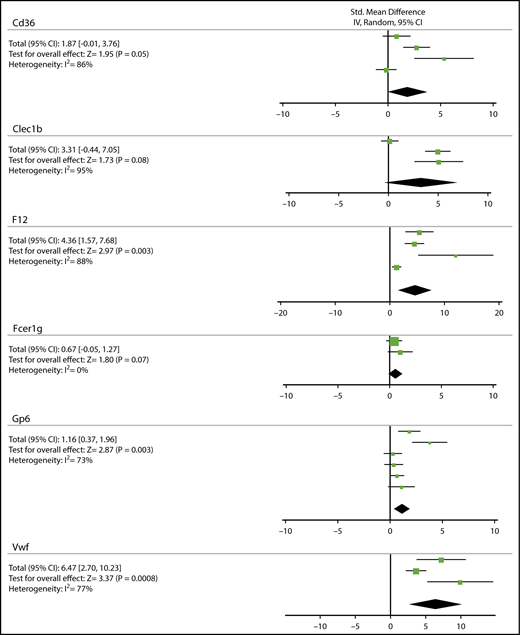
Meta-analysis on the contribution of Cd36, Clec1b, F12, Fcer1g, GP6, or Vwf mouse genes to prolongation of FeCl3-induced arterial thrombosis. Conventional meta-analysis assuming a standard random-effects model, to compare effects of genetic deficiency in Cd36, Clec1b, F12, Fcer1g, Gp6, or Vwf on prolongation time of arterial thrombus formation upon FeCl3 injury. Given per gene are: standardized mean difference with 95% confidence interval (CI), test for overall effect (Z), and heterogeneity index (I2). For details, see supplemental Data File 2.
Meta-analysis on the contribution of Cd36, Clec1b, F12, Fcer1g, GP6, or Vwf mouse genes to prolongation of FeCl3-induced arterial thrombosis. Conventional meta-analysis assuming a standard random-effects model, to compare effects of genetic deficiency in Cd36, Clec1b, F12, Fcer1g, Gp6, or Vwf on prolongation time of arterial thrombus formation upon FeCl3 injury. Given per gene are: standardized mean difference with 95% confidence interval (CI), test for overall effect (Z), and heterogeneity index (I2). For details, see supplemental Data File 2.
Study scaling and subdividing of parameters of arterial thrombus formation, thromboembolism, and tail-bleeding
As an alternative approach to compare and normalize published effects of gene inactivation on arterial thrombus formation and bleeding, we searched for a synthesis procedure that is condition independent. A simple method is to scale the published effect of genetic perturbation (eg, quantifiable difference between wild-type and genetically modified mice) on a 3-point or 5-point scale (Figure 1). Herein, the 3-point scale discriminates between a decrease and increase of the parameter determining the thrombotic process, whereas the 5-point scale further distinguishes between a moderate/strong decrease and increase. For convenience, we refer to the results of this scaling as “scores.”
For the database of 1431 studies (ie, those published before January 2018), we performed a consistency analysis to determine whether scores per class were comparable depending on the type of vascular bed, the injury trigger, and the parameter of measurement. The original papers were mined for quantitative data on arterial thrombosis, thromboembolism, or tail-bleeding (supplemental Methods). Weighing was not performed because of the similar sample size of studies and the lack of calibration options.
For studies on thrombus formation in vivo (class I) and in vitro (class III), we separated the published data into time-, mass-, or stability-dependent parameters. To accommodate different measurement outcomes, we further divided the mass-dependent parameters into 3 subparameters, that is, platelet adhesion, vessel occlusion, and thrombus size (supplemental Table 2).
Comparative analysis within studies of class I and class III, applied to the whole database, showed significant correlations between the 3 mass-dependent subparameters (class I: bi-biii, class III: gi-giii), at a 3-point and mostly also at a 5-point scale, with the Kendall τ ≥ 0.54 (P < .050) (supplemental Table 3A-B). This allowed combination of the subscores into 1 mass-dependent score per study. Subsequently, correlation analysis pointed to a high similarity of time-, mass-, and stability-dependent parameters, again within individual studies of the whole database (supplemental Table 3C-D). In class I, these parameters correlated with P < .001, except when comparing time and stability parameters at a 5-point scale (P = .226). In class III, with lower sample size, the time- and mass-dependent parameters correlated with P = .046 (3-point) or 0.072 (5-point); thrombus stability was hardly measured in class III. Together, the overall high consistency of scaling of time-, mass- and stability-dependent parameters made it possible to combine these per study into 1 mean synthesis score (3- and 5-point scale), as a calibrated outcome of the gene-modifying effect in that particular vascular bed and injury type. Studies of arterial thromboembolism (class II) and tail-bleeding times (class IV) were also scaled at 3- and 5-point.
Comparison of scaling for different experimental models of thrombus formation and thromboembolism
In 205 cases, mice of the same breeding were used in 1 paper for class I/II in vivo studies as well as for class III in vitro studies (supplemental Data File 1, same rows). This allowed us to directly compare the scores of such studies (supplemental Table 4). For the database as a whole, this resulted in significant correlations for the 3- and 5-point scaling of class I (P < .001; n = 151) and class II (P =.006 and .268; n = 14) studies, when compared with class III studies in the same paper. Hence, overall, the scaling of the in vitro thrombus formation appears to be in good agreement with that of in vivo arterial thrombosis.
Next, we compared per gene the results obtained for different vascular beds or injury types. Grouping the studies according to these variables (Figure 1; supplemental Table 2) revealed that, for class I studies, the mostly used vascular beds were carotid artery > mesentery > cremaster > aorta, whereas the most popular injury methods were FeCl3 > laser > photochemical > vessel ligation or compression > electrolytic injury (supplemental Figure 1A). For class II, the majority of papers applied injection of collagen/epinephrine, with fewer studies using tissue factor or reporting unprovoked thrombosis (supplemental Figure 1B). Concerning class III, collagen type I was mostly used as thrombogenic surface (supplemental Figure 1C). Class IV bleeding time studies were not subdivided.
For comparison of the different experimental approaches in the studies, groups in class I were combined with respect to vascular bed or injury mode. Regression analysis indicated significant correlations at 3-point and 5-point scales between the scores for carotis, mesentery, and cremaster injury, when averaged per gene (Table 1). Furthermore, we found highly significant (P < .001) correlations of mean scores per gene in class I for carotid or mesenteric injury (ie, collagen-dependent thrombosis models) with class III study scores (ie, collagen-induced thrombus formation in vitro). Markedly, the scaling of class I studies using cremaster injury (laser-induced, considered to be less collagen-dependent) again correlated with that of class III studies (P < .001, 3- and 5-point).
Correlations with scores per study in class I (in vivo thrombus formation) or class III (in vitro thrombus formation) for different vascular beds and different triggers
| Groups | N | 3-point | P | 5-point | P | |
|---|---|---|---|---|---|---|
| Kendall τ | Kendall τ | |||||
| Compared with carotis | (1-4) | |||||
| Mesentery | 6-8 | 53 | 0.80 | <.001 | 0.53 | <.001 |
| Cremaster | 9-11 | 27 | 0.89 | <.001 | 0.66 | <.001 |
| Compared with FeCl3 | (1, 6, 9) | |||||
| Laser | 8, 11 | 38 | 0.78 | <.001 | 0.50 | .001 |
| Photochemical | 2, 7, 10 | 27 | 0.67 | <.001 | 0.55 | .001 |
| Ligation and compression | 3, 5 | 23 | 0.59 | .003 | 0.52 | .005 |
| Compared with class III | (30) | |||||
| Carotis | 1-4 | 77 | 0.57 | <.001 | 0.49 | <.001 |
| Mesentery | 6-8 | 65 | 0.77 | <.001 | 0.65 | <.001 |
| Cremaster | 9-11 | 35 | 0.80 | <.001 | 0.64 | <.001 |
| FeCl3 | 1, 6, 9 | 99 | 0.67 | <.001 | 0.62 | <.001 |
| Laser | 8, 11 | 36 | 0.86 | <.001 | 0.56 | .001 |
| Photochemical | 2, 7, 10 | 15 | 0.26 | .325 | 0.46 | .056 |
| Ligation and compression | 3, 5 | 24 | 0.87 | <.001 | 0.70 | <.001 |
| Collagen/epinephrine | 20 | 14 | 0.71 | .006 | 0.25 | .286 |
| Groups | N | 3-point | P | 5-point | P | |
|---|---|---|---|---|---|---|
| Kendall τ | Kendall τ | |||||
| Compared with carotis | (1-4) | |||||
| Mesentery | 6-8 | 53 | 0.80 | <.001 | 0.53 | <.001 |
| Cremaster | 9-11 | 27 | 0.89 | <.001 | 0.66 | <.001 |
| Compared with FeCl3 | (1, 6, 9) | |||||
| Laser | 8, 11 | 38 | 0.78 | <.001 | 0.50 | .001 |
| Photochemical | 2, 7, 10 | 27 | 0.67 | <.001 | 0.55 | .001 |
| Ligation and compression | 3, 5 | 23 | 0.59 | .003 | 0.52 | .005 |
| Compared with class III | (30) | |||||
| Carotis | 1-4 | 77 | 0.57 | <.001 | 0.49 | <.001 |
| Mesentery | 6-8 | 65 | 0.77 | <.001 | 0.65 | <.001 |
| Cremaster | 9-11 | 35 | 0.80 | <.001 | 0.64 | <.001 |
| FeCl3 | 1, 6, 9 | 99 | 0.67 | <.001 | 0.62 | <.001 |
| Laser | 8, 11 | 36 | 0.86 | <.001 | 0.56 | .001 |
| Photochemical | 2, 7, 10 | 15 | 0.26 | .325 | 0.46 | .056 |
| Ligation and compression | 3, 5 | 24 | 0.87 | <.001 | 0.70 | <.001 |
| Collagen/epinephrine | 20 | 14 | 0.71 | .006 | 0.25 | .286 |
Indicated are correlations across genes for similar genetically modified mice, used for studies in multiple classes, when scored on 3-point or 5-point scales. N refers to number of comparisons (genes) made. Comparison for indicated groups (injury modes, in italic) are indicated relative to carotid models (groups 1-4), FeCl3 injury models (groups 1, 6, 9), or in vitro studies (group 30).
When comparing different injury modes in class I, the scaling per gene for FeCl3 studies correlated significantly (P ≤ .005, 3- and 5-point) with that for laser, photochemical, or ligation/compression injuries (Table 1). Furthermore, class III scores of collagen-dependent thrombus formation were related on a 3-point scale (P =.006, low n = 14) but not on a 5-point scale (P = .286) with class II scores (thromboembolism with frequent collagen/epinephrine injection). Taken together, this analysis revealed that, overall, the scaling for classes I, II, and III gave similar outcomes for effects of a certain gene deficiency, irrespective of the arterial vascular bed, injury mode, injection of collagen, or in vitro testing.
Synthesis approach of scaled studies to compare roles of mouse genes in thrombus formation, thromboembolism, and hemostasis
Considering that the 5-point scale (classes I-III) is more discriminative than the 3-point scale, we used the former for comparative analyses of the 1431 studies. As a first approach, mean scaled scores were calculated per class and gene. For 409 genes, this resulted in information on 277, 79, and 161 genes for classes I, II, and III, respectively (Figure 3A). For bleeding times (class IV), information was collected for 314 genes (Figure 3A). First analysis across all genes revealed that a gene defect with a negative score for arterial thrombosis was more frequently (69%) accompanied with prolonged bleeding than unchanged bleeding (Figure 3C).
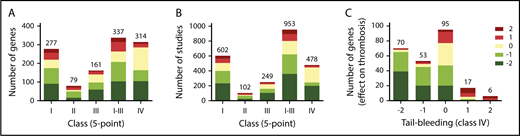
Score distribution patterns of studies and mouse genes with increased or decreased thrombosis tendency or bleeding. Indicated are score distributions per class of numbers of mouse genes (A) or numbers of studies (B). Classes I-III refer to arterial thrombosis in vivo, systemic thromboembolism, and thrombus formation in vitro, respectively; class IV refers to tail-bleeding times. In all cases, scoring was at 5-point scale. Studies using mice with noncomparable genetics (eg, gain-of-function mutations) were not included. (C) Numbers of mouse genes with altered scores in classes I-III (5-point scale), in combination with prolonged (−2/−1), unchanged (0), or shortened (+1/+2) tail-bleeding time (class IV).
Score distribution patterns of studies and mouse genes with increased or decreased thrombosis tendency or bleeding. Indicated are score distributions per class of numbers of mouse genes (A) or numbers of studies (B). Classes I-III refer to arterial thrombosis in vivo, systemic thromboembolism, and thrombus formation in vitro, respectively; class IV refers to tail-bleeding times. In all cases, scoring was at 5-point scale. Studies using mice with noncomparable genetics (eg, gain-of-function mutations) were not included. (C) Numbers of mouse genes with altered scores in classes I-III (5-point scale), in combination with prolonged (−2/−1), unchanged (0), or shortened (+1/+2) tail-bleeding time (class IV).
Subsequently, for the 35 best-investigated genes (n ≥ 6 studies across classes I-III, 65% collagen-dependent models), we evaluated the distribution of study scores in more detail (Figure 4A). We considered these scores as discretized ordinal variables with defined ranking; hence, these could be treated as interval variables. This allowed calculation of 95% confidence intervals and P values, while keeping the rounded-off mean scaling per gene (Figure 4B).
![Figure 4. Consistency analysis for the most frequently studied genes on effects on arterial thrombosis. (A) Summation of scores per study for 35 indicated genes in classes I-III with n ≥ 6. Data obtained from 312 studies (65% collagen-dependent models, 15% laser models, 7% photochemical injury). (B) Mean scores per gene with 95% CI. (C) Calculated CP per gene [1 −log(P)]. Note that a CP value > 2.30 corresponds to a P < .05, and to 95% CI values not including zero.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/132/24/10.1182_blood-2018-02-831982/6/m_blood831982f4.png?Expires=1767733560&Signature=Zv-dCtna2lHj3iby393SiOTKwyGJfaWaRb7k9Wbt2P5KPwVsrPaTTwP9xPKxOOFvU3PqVQogRBhcsjdaRjwmLrA1I4tSCSDDwPIivSXWDeQdC9gy6soMT5EM1R6G~5q54e7yBCnU0CjsuPaibkODe9LZfV4ErOscuqH6vZkmffOBraqTzgIJWX~wPtEvcabNmwqV8965a0NM75ozXTgUOBh4Yzf5nOl~nicUBNsaO302G5CDwJ034VT6MCneqlQQCt1OlfNTqdq5TSTYYlHrcIFnM0jmAKZzcemiLxBq-jQUZoivkLhNEnQcf29zY3p0Oesq36XVCE9BWmUVMpeDsw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Consistency analysis for the most frequently studied genes on effects on arterial thrombosis. (A) Summation of scores per study for 35 indicated genes in classes I-III with n ≥ 6. Data obtained from 312 studies (65% collagen-dependent models, 15% laser models, 7% photochemical injury). (B) Mean scores per gene with 95% CI. (C) Calculated CP per gene [1 −log(P)]. Note that a CP value > 2.30 corresponds to a P < .05, and to 95% CI values not including zero.
Consistency analysis for the most frequently studied genes on effects on arterial thrombosis. (A) Summation of scores per study for 35 indicated genes in classes I-III with n ≥ 6. Data obtained from 312 studies (65% collagen-dependent models, 15% laser models, 7% photochemical injury). (B) Mean scores per gene with 95% CI. (C) Calculated CP per gene [1 −log(P)]. Note that a CP value > 2.30 corresponds to a P < .05, and to 95% CI values not including zero.
A standard method to assess distribution profiles of interval variables does not exist. Based on P values obtained from normalized data (per gene, see “Methods”), we therefore defined a CP as 1 − log(P), reflecting the uniformity and strength of the pooled study scores. Accordingly, we considered that a mean nonzero scaling with CP > 2.30 (corresponding to P < .05) consistently points to an anti- or prothrombotic propensity.
Following this synthesis approach it appeared that, for the best studied genes, 14 genes with a mean −2 score gave a CP > 2.30 (in decreasing order: Gp6, Fcer1g, F12, Vwf, Itgb3, Rasgrp2, P2ry12, Pik3cb, F8, Pdia3 > Tln1, F2, F9 > Lcp2) (Figure 4C). Furthermore, 13 of 17 genes with a mean −1 score had a CP > 2.30 (Ptprj, F11, P2ry1 > F2rl3 > Orai1, Stim1, Serpine1, Gp1bb, Pld1 > Klkb1, Itga2, Selp, Clec1b). Regarding the “prothrombotic” gene with mean score +1, Adamts13 gave a CP > 2.30. Negative scores of the genes Cd36, Itgb1, Jak2, and Vtn did not reach significance (Figure 4C).
The suitability of this synthesis approach prompted us to generate mean scores and CP values for all genes. In this way, it is possible to compare, across all genes, not only the overall effect size (5-point scale) but also the consistency of the reported data of a gene defect, regarding both arterial thrombosis/thromboembolism (class I-III) and a bleeding tendency (class IV). Supplemental Data File 3 provides an overview of all gene defects (or pharmacological intervention), regarding mean class scores, CP values, and a short description of altered platelet, coagulant, or vascular function.
For 60 genes with CP > 2.30 (P < .05) and an arbitrary cutoff of n ≥ 3 studies, the results are summarized in Table 2, separating mice with an “antithrombotic” phenotype (negative class I-III scores) and a normal or prolonged bleeding time. For 17 genes, mostly encoding platelet receptor or signaling proteins or plasma coagulation proteins, there appears to be consistent evidence for a role in arterial thrombosis but not hemostasis. For 42 genes, encoding proteins with similar functions, the scaled data points to a contribution to both thrombosis and hemostasis. In 1 case (deficiency in Ceacam1/2), a prothrombotic phenotype is accompanied with prolonged bleeding, possibly due to vascular changes.
Mouse and orthologous human genes with consistent contribution to arterial thrombosis with(out) altered bleeding
| Altered phenotype | Mouse deficiency | Human ortholog |
|---|---|---|
| Antithrombotic, no prolonged bleeding | ||
| Plasma coagulation | F11, F12 | F11*, F12* |
| Plasma fibrinolysis | Serpine1 | SERPINE1 |
| Platelet membrane | Dusp3, Gp6, Itga6, P2rx1, Selp | DUSP3, GP6*, ITGA6, P2RX1, SELP |
| Platelet signaling | Csnk2b,Pik3c3, Pik3cg, Pld1, Ppia, Prkca, Rhog, S100a9, Sgk1 | CSNK2B, PIK3C3, PIK3CG, PLD1, PPIA, PRKCA, RHOG, S100A9, SGK1 |
| Antithrombotic, prolonged bleeding | ||
| Coagulation | F2, F8, F9, Klkb1, Vwf | F2*, F8, F9, KLKB1, VWF* |
| Platelets | ||
| Membrane proteins | Ano6, Clec1b, F2rl3, Fcer1g, Fermt3, Gna13, Gnaq, Gp1ba, GP1bb, Itga2, Itgb3, Mertk, Orai1, P2ry1, P2ry12, Pdia3, Pdia4, Ptprj, Stim1, Trpm7, Tspan32 | ANO6, CLEC1B, F2RL3*, FCER1G, FERMT3, GNA13, GNAQ, GPIBA*, GPIBB, ITGA2, ITGB3*, MERTK, ORAI1, P2RY1, P2RY12*, PDIA3, PDIA4, PTPRJ, STIM1, TRPM7, TSPAN32 |
| Signaling | Csk, Map3k5, Mapk8, Mapk14, Pdpk1, Pik3cb, Plcg2, Rac1, Rasgrp2, Tln1 | CSK, MAP3K5, MAPK8, MAPK14, PDPK1, PIK3CB*, PLCG2, RAC1, RASGRP2, TLN1 |
| Secretion/activation | Nbeal2, Kcnip3, Mmp2 | NBEAL2, KCNIP3, MMP2 |
| Vessel wall | Bambi, Bdkrb2,Mfap2 | BAMBI, BDKRB2, MFAP2 |
| Not antithrombotic, prolonged bleeding | ||
| Vessel wall | Ceacam1/2 | CEACAM1 |
| Altered phenotype | Mouse deficiency | Human ortholog |
|---|---|---|
| Antithrombotic, no prolonged bleeding | ||
| Plasma coagulation | F11, F12 | F11*, F12* |
| Plasma fibrinolysis | Serpine1 | SERPINE1 |
| Platelet membrane | Dusp3, Gp6, Itga6, P2rx1, Selp | DUSP3, GP6*, ITGA6, P2RX1, SELP |
| Platelet signaling | Csnk2b,Pik3c3, Pik3cg, Pld1, Ppia, Prkca, Rhog, S100a9, Sgk1 | CSNK2B, PIK3C3, PIK3CG, PLD1, PPIA, PRKCA, RHOG, S100A9, SGK1 |
| Antithrombotic, prolonged bleeding | ||
| Coagulation | F2, F8, F9, Klkb1, Vwf | F2*, F8, F9, KLKB1, VWF* |
| Platelets | ||
| Membrane proteins | Ano6, Clec1b, F2rl3, Fcer1g, Fermt3, Gna13, Gnaq, Gp1ba, GP1bb, Itga2, Itgb3, Mertk, Orai1, P2ry1, P2ry12, Pdia3, Pdia4, Ptprj, Stim1, Trpm7, Tspan32 | ANO6, CLEC1B, F2RL3*, FCER1G, FERMT3, GNA13, GNAQ, GPIBA*, GPIBB, ITGA2, ITGB3*, MERTK, ORAI1, P2RY1, P2RY12*, PDIA3, PDIA4, PTPRJ, STIM1, TRPM7, TSPAN32 |
| Signaling | Csk, Map3k5, Mapk8, Mapk14, Pdpk1, Pik3cb, Plcg2, Rac1, Rasgrp2, Tln1 | CSK, MAP3K5, MAPK8, MAPK14, PDPK1, PIK3CB*, PLCG2, RAC1, RASGRP2, TLN1 |
| Secretion/activation | Nbeal2, Kcnip3, Mmp2 | NBEAL2, KCNIP3, MMP2 |
| Vessel wall | Bambi, Bdkrb2,Mfap2 | BAMBI, BDKRB2, MFAP2 |
| Not antithrombotic, prolonged bleeding | ||
| Vessel wall | Ceacam1/2 | CEACAM1 |
Mouse genes are listed with n ≥ 3 scaled studies (5-point) and CP values of >2.30 (P < .05). Phenotype classification was according to reported changes regarding plasma, platelet, or vascular phenotypes. Bold indicates mouse deficiency resulting in a highly prolonged bleeding (class IV, score −2).
Human orthologs targeted in current clinical practice or trials.
When comparing the human orthologs of these 42 genes, it appears that mutations are often associated with bleeding events,19 especially for those corresponding to mouse genes with a −2 bleeding score (Table 2). These include F8, F9, VWF, FERMT3, GPIBA, GPIBB, ITGB3, P2RY1, P2RY12, RASGRP2, TLN1, and NBEAL2. Interestingly, for several other genes (eg, GNA13, MAP3K3, KCNIP3), evidence in humans of a possible role in human hemostasis is still unknown. We note here that, given the low prevalence of inherited Mendelian bleeding disorders, very little is known of a protective role in thrombosis in this group of rare patients with hemostatic insufficiencies. The mouse data would predict such a role.
Antagonists for several human orthologous proteins are already in use for patient treatment (thrombin inhibitors, F2; integrin αIIbβ3 antagonists, ITGB3; P2Y12 antagonists, P2RY12) or are studied in clinical trials (inhibitors of: factor XI and XII, F11, F12; glycoprotein VI, GP6; glycoprotein Ib-VWF, GPIBA, von Willebrand factor [VWF]; phosphoinositide 3-kinase-β, PIK3CB).
Reactome biological pathway analysis of (modifying) genes in arterial thrombosis and hemostasis
To translate the scoring per mouse gene/protein to human pathophysiology, we first performed an overrepresentation analysis using the Reactome database. This knowledge base of biological pathways is elaborated for megakaryocytes, platelets, and the coagulation system.20 Pathway analysis for 405 scored genes/proteins indicated that a substantial part of the Reactome-registered hemostatic system was covered (148 of 806 proteins) (supplemental Data File 5). Well-covered pathways included platelet activation, platelet/neutrophil degranulation, and fibrin clot formation. Several unexpected pathways were also detected, for example, linked to cancer (see supplemental Data File 5). Additional Reactome attribute analysis, taking into account the scores per gene/protein, revealed pathways where mean scores for (modifying) proteins in class I-III were less negative than those for proteins in class IV (supplemental Data File 5). This concerned pathways of 12/15-eicosatetraenoic synthesis, matrix metalloproteinases, and common fibrin clot formation, suggesting that these may play a more prominent role in hemostasis. Conversely, mean scores for proteins of Toll-like receptor pathways were more negative in class I-III than those for proteins in class IV, suggesting a more prominent role in thrombosis (supplemental Data File 5).
Network of genes and proteins implicated in arterial thrombus formation
To further exploit the data set, a Cytoscape network was constructed with human protein orthologs of 267 genes with modifying effects on arterial thrombosis (class I-III). Using these as primary baits, a network was obtained retrieving 2679 first-order interactors (novel nodes) and 19 721 interactions (edges) (Figure 5A). The expanded network of protein interactions in thrombosis and hemostasis (PITH) was used for further exploration of new (patho)physiological pathways (supplemental Data File 6).
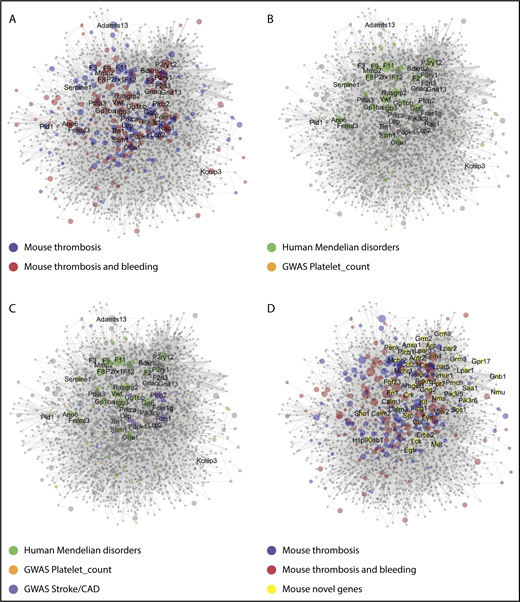
Network of protein interactions in thrombosis and hemostasis. Network built in Cytoscape for 275 bait proteins, corresponding to mouse genes with modifying effect on arterial thrombosis of hemostasis. The final network has 267 core genes (bait nodes), 2679 new nodes, connected by 19721 interactions (edges). Of the edges, 15 419 are derived from Reactome, 4299 from Intact, and 3 from manual assignment. Size of nodes is set as 1 per default, or as CP value. (A) Network visualization with color-coded bait nodes: proteins modifying thrombosis (blue) or thrombosis plus bleeding (red). Names are listed of 40 mouse genes with highest CP values. (B) Network visualization with color-coded proteins linked to rare Mendelian disorders of human platelet or coagulant function (green, n = 54); proteins linked to high-association genes in GWAS of platelet count and volume (orange, n = 21). (C) Network visualization with additional purple-coded proteins linked to a recent GWAS of stroke (n = 11) and coronary artery disease (n = 40). (D) Network visualization emphasizing 50 novel nodes with largest number of edges (≥25, yellow). Attribute lists are given in supplemental Data File 6. The full network, containing gene-expression levels in human megakaryocytes33 and other annotation features, is available in Cytoscape format upon request. For enlarged panels, see supplemental Figure 3.
Network of protein interactions in thrombosis and hemostasis. Network built in Cytoscape for 275 bait proteins, corresponding to mouse genes with modifying effect on arterial thrombosis of hemostasis. The final network has 267 core genes (bait nodes), 2679 new nodes, connected by 19721 interactions (edges). Of the edges, 15 419 are derived from Reactome, 4299 from Intact, and 3 from manual assignment. Size of nodes is set as 1 per default, or as CP value. (A) Network visualization with color-coded bait nodes: proteins modifying thrombosis (blue) or thrombosis plus bleeding (red). Names are listed of 40 mouse genes with highest CP values. (B) Network visualization with color-coded proteins linked to rare Mendelian disorders of human platelet or coagulant function (green, n = 54); proteins linked to high-association genes in GWAS of platelet count and volume (orange, n = 21). (C) Network visualization with additional purple-coded proteins linked to a recent GWAS of stroke (n = 11) and coronary artery disease (n = 40). (D) Network visualization emphasizing 50 novel nodes with largest number of edges (≥25, yellow). Attribute lists are given in supplemental Data File 6. The full network, containing gene-expression levels in human megakaryocytes33 and other annotation features, is available in Cytoscape format upon request. For enlarged panels, see supplemental Figure 3.
To relate to human diseases, the network was linked to several large-scale human genetic studies. As nodes, we could trace 54 genes of, in total, 72 genes linked to rare human Mendelian disorders of platelet or coagulation function, obtained in the National Institute for Health Research (NIHR) BioResource Bleeding, Thrombotic and Platelet Disorders cohort,21 and 21of 68 genes from a genome-wide association study (GWAS) of platelet count and volume22 (Figure 5B). The latter set encompassed 147 DNA variants in the network of 647 variants obtained in a more recent GWAS of platelet quantitative traits (supplemental Data File 6).23 Regarding arterial thrombosis, 11 genes were identified in 39 loci from a GWAS of stroke,24 and 40 genes in 64 loci from a GWAS of coronary artery disease25 (Figure 5C). Thus, although the network was constructed from only 267 core proteins/genes, it covered substantial gene sets identified in GWAS and related studies.
Subsequently, we evaluated node connections and node centrality characteristics, such as proximity to other nodes (high for core nodes) and tendency to subclustering (low for core nodes) (supplemental Data File 6). Messenger RNA (mRNA) expression levels in human megakaryocytes were determined for the network (supplemental Figures 4 and 5A). For the novel nodes, it appeared that a substantial part of the corresponding genes were transcribed in megakaryocytes (supplemental Figures 4A and 5B), and that most of these had 1 to 4 connections with 1 or more modifying proteins in the network (supplemental Figure 4B). Subsequently, we isolated the top 50 most connected novel proteins (Figure 5D), revealing not only some known proteins (Cbl, Fyn, Lck) but also proteins of novel pathways (Anxa, Grm, Lpar, Nmur family members).
Altered thrombus formation of mice deficient in newly identified genes
Taking into account the node characteristics, the PITH network was subsequently used to identify novel mouse genes with a possible role in arterial thrombosis. From the list of novel nodes with edges to modifying genes, we screened 2 groups of knockout mice for in vitro arterial thrombus formation (class III), which had not been evaluated yet in this way. One group came from the extensive Mouse Genetics breeding program of the Wellcome Sanger Institute in Cambridge (United Kingdom). These mice carried a homozygous deficiency in Bnip2 (regulating cell death), Dlg4 (membrane-associated guanylate kinase clustering signaling proteins), Grm8 (Gi-coupled metabotropic glutamate receptor 8), Ifnar1 (interferon receptor signaling via the JAK/STAT pathway), or Vps13a (involved in protein cycling through the trans-Golgi network) (Figure 6A). Mice deficient in 2 of 4 genes with high mRNA expression in reference megakaryocytes (Figure 6B), that is, Ifnar1 and Vps13a but not Bnip2 and Dlg4, gave a significant reduction in arterial thrombus formation (score −2). These findings are corroborated by recent papers showing that interferon I (ligand of Ifnar1 transcript) regulates platelet count and function,26 and that gene defects in human VPS13A are associated with chorea-acanthocytosis, where patients carry platelets with impaired granule secretion.27 In accordance with low mRNA expression in reference megakaryocytes, Grm8 deficiency did not affect thrombus formation (score 0).
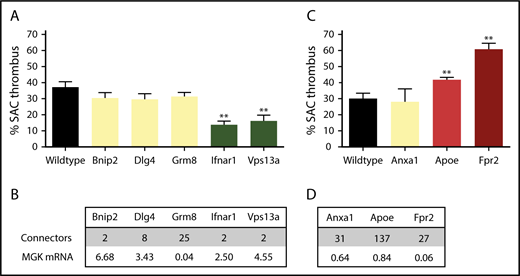
Role of 8 novel mouse genes in arterial thrombus formation, based on PITH network. Mice with homozygous deficiency in indicated genes were tested for arterial thrombus formation (class III). Data represent surface area coverage (SAC) of platelet thrombi on collagen. (A-B) Testing of 5 genes/proteins selected from the PITH network with suspected thrombosis phenotype. Mean plus or minus standard error (SE; n = 6-17); **P < .01. (C-D) Testing of 3 genes/proteins identified from PITH with suspected gain-of-function defect related to atherothrombosis. Mean plus or minus SE (n = 6-18); **P < .01. (B,D) Connecting edges in PITH network, and relative mRNA expression in human megakaryocytes (log2 scale). MGK, megakaryocyte.
Role of 8 novel mouse genes in arterial thrombus formation, based on PITH network. Mice with homozygous deficiency in indicated genes were tested for arterial thrombus formation (class III). Data represent surface area coverage (SAC) of platelet thrombi on collagen. (A-B) Testing of 5 genes/proteins selected from the PITH network with suspected thrombosis phenotype. Mean plus or minus standard error (SE; n = 6-17); **P < .01. (C-D) Testing of 3 genes/proteins identified from PITH with suspected gain-of-function defect related to atherothrombosis. Mean plus or minus SE (n = 6-18); **P < .01. (B,D) Connecting edges in PITH network, and relative mRNA expression in human megakaryocytes (log2 scale). MGK, megakaryocyte.
The second group was provided by breeding programs of the Institute for Cardiovascular Prevention in Munich (Germany). This included animals with a homozygous deficiency in apolipoprotein E (Apoe, controlling the catabolism of lipoprotein triglycerides; affecting thrombosis in vivo, but not yet evaluated in class III), formyl peptide receptor 2 (Gi protein–coupled Fpr2), or annexin A1 (Anxa1, FPR2 ligand) (Figure 6C). These genes have been implicated in arteriosclerosis, and appear with multiple edges in the PITH network. Transcripts are expressed at medium to high levels in the liver,28 but only Anxa1 and Apoe transcripts are detectable at low levels in megakaryocytes. We found that deficiencies in Apoe and Fpr2, but not Anxa1, positively modified thrombus formation (Apoe−/−, score +1 and Fpr2−/−, score +2) (Figure 6D). This implies that the prothrombotic role of FPR2 is independent of its proposed ligand ANX1. It appeared that 4 from the 8 novel genes with a predicted contribution to arterial thrombus formation showed a phenotype in thrombus formation (Ifnar1, Vps13a, Apoe, Fpr2).
Discussion
Formal meta-analyses were performed to compare studies investigating consequences of genetic deficiency for experimental arterial thrombosis. The analyses were greatly impaired by high heterogeneity, as a consequence of small sample size (typically n = 6-12) and differences in experimental setup. To overcome these limitations, we developed an alternative synthesis approach, in which mouse studies of arterial thrombosis, thromboembolism, in vitro thrombus formation, or bleeding were scaled, regardless of the thrombosis model and output parameters, that is, features that greatly differ from laboratory to laboratory.10
Using a uniform normalization or scoring approach, we compared in total 1514 individual studies, investigating deficiencies in 431 mouse genes. Statistical analysis to compare 3-point and 5-point scores for different modes of thrombus induction, vascular beds, and outcome parameters underlined the overall usefulness of a universal normalization procedure. It, however, also revealed subtle differences between established collagen-dependent models (ie, FeCl3 or ligation/compression injury) and thrombosis models that were less collagen-dependent (laser or photochemical injury). This is in agreement with papers indicating that the laser model is more dependent on tissue factor activity and/or mild endothelial damage, rather than collagen exposure, although this may depend on the severity of vascular trauma.29-31 Regression analysis pointed to lower significance when comparing high vs low collagen-dependent models.
Based on the statistical analysis of the collated data, it can be recommended for future thrombosis studies in mice to combine the results of the most commonly used models (FeCl3, ligation and laser injury of the carotid artery) in combination with multiple outcome measurements (ie, not only time to occlusion). Taken together, the overall agreement in scoring scales, across main experimental variables, indicates that, in general, the effect of gene modification is larger than the variation caused by differences in methodology or measurement. In a next step, we accumulated the study scores per gene (treated as interval variables to allow statistical comparison) to calculate the CP as a measure of both the similarity and the number of evaluated studies.
This approach provided a valuable list of 60 consistently studied mouse genes (n ≥ 3, CP > 2.30), which in majority encode coagulation, platelet membrane, or signaling proteins contributing to thrombosis and/or hemostasis (Table 2). Together with mouse models of human disease (eg, Ano6, F8, Itgb3, Nbeal2, Vwf), this list also contains many unexpected genes, where the relation to human disease still needs to be explored. For these 60 mouse genes/proteins, an overall high homology was present with the human orthologs (supplemental Table 7). Given that only a few drug targets are currently used for thrombosis treatment, this synthesis approach reveals several candidate proteins with a possible role in arterial thrombosis without bleeding. These include proteins that modulate functions of platelets, lipids, coagulation, or fibrinolysis, as well as the vasculature.
In addition, our list contains only a few thrombosis-suppressive genes (eg, encoding for anticoagulant, fibrinolytic, or vascular proteins). For several of these, in humans, deficiency is linked to an increased risk of (arterial) thrombosis (protein C, S, antithrombin). It will be important now to also investigate the thrombosis risk in humans for the other mouse genes with positive thrombosis scaling. On the other hand, human genetics can also be less straightforward, as exemplified by the observation that a mutation of the F5 (factor V) gene linked to altered binding of anticoagulant factors leads to a bleeding disorder,32 whereas the factor V Leiden allele of F5 increases the risk of thrombosis.
For a larger set of 275 modifying mouse genes, information (n ≥ 1, CP ≥ 1) was present regarding a positive or negative role in arterial thrombosis (supplemental Data File 3). This synthesis approach thus may serve as an alternative for meta-analyses on mouse studies outside of the field of thrombosis and hemostasis. Our synthesis approach also indicates that, for genes implicated in platelet function, measurement of thrombus formation using flow chambers in vitro can serve as a proxy test for assessment of (collagen-dependent) thrombus formation in vivo. Also, this supports implementation of the 3 Rs by: (i) refining in vitro procedures limiting the need for in vivo experiments; (ii) reducing numbers of animals, given the relatively small blood volumes needed for in vitro tests, and (iii) ultimately replacing animal by human blood.
The PITH network revealed a wealth of candidate proteins and pathways possibly regulating arterial thrombus formation. Guided by node and centrality characteristics of the network, we could identify 4 novel mouse genes with a role in thrombus formation. Linking the network to large human databases of genetics of hemostasis and arterial thrombosis (supplemental Data File 6), mouse-human homology (supplemental Data File 4), and online Mendelian inheritance in man (OMIM) information (supplemental Table 6) confirms its translational relevance. Hence, the PITH network can be used for setting priors when analyzing whole genome-sequencing efforts for patients with rare inherited bleeding, thrombotic, or platelet disorders of unknown molecular etiology. It must be noted though that not all genes included in the PITH network are expressed in megakaryocytes and platelets, that functional redundancy can exist between protein isoforms, and that such redundancy can be different across species. Hence, single-gene or protein deficiencies may not necessarily cause a thrombotic or hemorrhagic phenotype.
The present synthesis approach also has limitations. One restriction is the small number of mostly heterogeneous, low-powered studies per gene. The present synthesis approach aimed to integrate heterogeneous studies. Correlation analysis indicated consistency between the scores of groups and classes, indicating that the grouping and synthesis approach is justified. Another limitation is that the compiled mouse data include a relatively large set of genes that, in humans, are linked to known inherited bleeding disorders, whereas genes with a role in the vascular component of thrombotic disorders are underrepresented. A limitation of the collagen-based in vitro measurements of thrombus formation is that anti/prothrombotic changes due to altered coagulation, fibrinolysis, or vascular dysfunction are not easily picked up.
In summary, this paper demonstrates that a novel synthesis approach based on scaling of distinct parameters of thrombus formation can integrate the effects of multiple small-size mouse studies to identify: (i) key genes and pathways of thrombotic processes in humans and (ii) new molecular targets for antithrombotic therapy.
The full-text version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
S.M.d.W., J.M.E.M.C., and J.W.M.H. have received funding from the Cardiovascular Center Maastricht, ZonMW (MKMD 114021004), the Interreg V Euregio Meuse-Rhine program (Poly-Valve), and The Netherlands Organization for Scientific Research (Nederlandse Organisatie voor Wetenschappelijk Onderzoek Vidi 91716421; J.M.E.M.C.). Research in the Ouwehand laboratory was supported by program grants from the National Institute for Health Research (M.K., S.M., and W.H.O.) and the British Heart Foundation under numbers RP-PG-0310-1002 and RG/09/12/28096; the laboratory also received funding from National Health Service (NHS) Blood and Transplant. M.K. was supported by Marie Curie funding from the NETSIM FP7 program, funded by the European Commission. O.S. received funding from the German Research Foundation (SO876/6-1, SFB914 B08, SFB1123 A06 and B05) and Nederlandse Organisatie voor Wetenschappelijk Onderzoek (91712303). S.J. was supported by the National Human Genome Research Institute at the National Institutes of Health (U41 HG003751).
Authorship
Contribution: C.C.F.M.J.B., S.M., and S.M.d.W. performed experiments, analyzed and interpreted data, and wrote the paper; M.A.H.F. and J.M.E.M.C. analyzed and interpreted data and revised the manuscript; D.J.A., J.-W.N.A., R.R.-S., O.S., C.W., and J.K.W. provided essential tools and revised the paper; S.J., L.G., M.K., N.J.A.M., and F.S. analyzed data; M.H.P. interpreted data and revised the manuscript; and W.H.O. and J.W.M.H. designed research, analyzed and interpreted data, and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Johan W. M. Heemskerk, Department of Biochemistry, CARIM, Maastricht University, PO Box 616, 6200 MD Maastricht, The Netherlands; e-mail: jwm.heemskerk@maastrichtuniversity.nl; or Willem H. Ouwehand, Department of Haematology, University of Cambridge & Wellcome Sanger Institute and NHS Blood and Transplant, Cambridge Biomedical Campus, Cambridge, CB2 0PT United Kingdom; e-mail: who1000@cam.ac.uk.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal