

In this issue of Blood, show that PU.1 autoregulation and the looping factor Lim domain–binding 1 (LDB1) are required for dynamic chromosome architecture remodeling involving PU.1 during myeloid differentiation, which is impaired in acute myeloid leukemia (AML).1
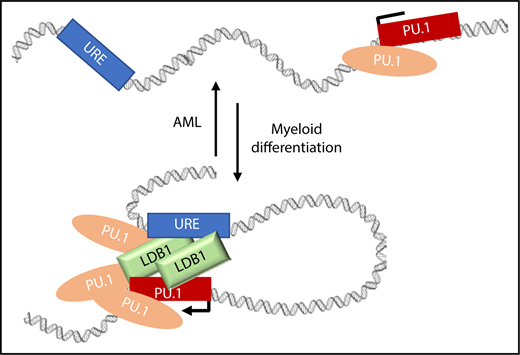
Model depicting how PU.1 and LDB1 could support URE contacts with the PU.1 gene in normal and abnormal situations. LDB1 and PU.1 accumulate at the PU.1 promoter and at the URE supporting contacts during myeloid differentiation in parallel with increasing PU.1 expression. In AML, PU.1 expression is low, and these long-range contacts are reduced. One testable explanation is that PU.1 and LDB1 occupancy in the locus is reduced in AML and that this results in loss of contacts between the URE enhancer and PU.1.
Model depicting how PU.1 and LDB1 could support URE contacts with the PU.1 gene in normal and abnormal situations. LDB1 and PU.1 accumulate at the PU.1 promoter and at the URE supporting contacts during myeloid differentiation in parallel with increasing PU.1 expression. In AML, PU.1 expression is low, and these long-range contacts are reduced. One testable explanation is that PU.1 and LDB1 occupancy in the locus is reduced in AML and that this results in loss of contacts between the URE enhancer and PU.1.
The eukaryotic genome is organized at successive levels into chromosome territories, active or repressed transcriptional compartments (A and B, respectively), and topologically associating domains (TADs) that are shared among cell types and even among different species.2 DNA sequences within TADs interact with each other and, for the most part, not with sequences outside the TAD boundaries. Enhancers and their target genes overwhelmingly occupy the same TAD. Thus, within TADs transcriptomic specificity is achieved that forms the basis of development and differentiation of unique cell lineages and tissue types.
PU.1 (encoded by the Spi1 gene) is an E-twenty-six (ETS)-family transcription factor that is critical for development of almost all blood cell lineages. Downregulation of PU.1 in early hematopoietic progenitors is necessary for their differentiation into the T-lymphoid and erythroid lineages. In contrast, PU.1 upregulation must occur for myeloid cell differentiation. In AML, PU.1 expression is commonly downregulated, consistent with the idea that reduction of PU.1 blocks myeloid differentiation.
Expression of PU.1 is regulated by long-range interaction with an enhancer, the upstream regulatory element (URE), located at −14 kb with respect to the transcription start site of the gene in mice (−17 kb in human).3 The PU.1 locus resides in a SubTAD, a defined TAD substructure, according to published Hi-C chromatin conformation capture data for human monocyte THP-1 cells.4 How the URE enhancer functions during differentiation of myeloid cells and development of myeloid malignancies is a subject of considerable interest. Disruption of three-dimensional (3D) chromosome architecture is an emerging disease mechanism, and, thus, manipulation of chromosome architecture presents a novel opportunity for therapeutic intervention.
Schuetzmann et al compared the 3D organization of the PU.1-encoding locus in normal human monocytes and in AML cells, using high-resolution circularized chromosome conformation capture followed by deep sequencing (4C-seq). They found that the URE enhancer and gene were in physical proximity in monocytes. Strikingly, the contacts were diminished in leukemic blast cells of 5 different AML patients coordinately with decreased expression of PU.1. The results establish a correlative, if not a causal, relationship between the PU.1/URE contacts and PU.1 expression in monocytes and AML cells.
To investigate further, the authors induced THP-1 cells to differentiate into macrophage-like cells with phorbol 12-myristate 13-acetate and observed acquisition of URE contacts by PU.1 in concert with upregulation of expression. In support of PU.1 autoregulation, they found several sites of PU.1 binding within the locus SubTAD by chromatin immunoprecipitation sequencing (ChIP-seq). Contact between the PU.1 gene and the URE occurred in parallel with an increase in PU.1 binding at the gene promoter and at the URE. The authors next engineered THP-1 cells with a doxycycline (dox)–inducible knockdown of PU.1. By reducing PU.1 expression either before or after induction of differentiation, they found that PU.1 was required to initiate the PU.1/URE gene contact but was dispensable after the contact was established.
This result raised the question of what was involved in maintaining the contact. LDB1 was investigated based on its role in enhancer looping in erythroid cells. LDB1 is a widely expressed nuclear protein that was originally identified as a cofactor for LIM-homeodomain and LIM-only proteins that have fundamental roles in development. LDB1-null mice die at embryonic day 8.5 with severe developmental defects in multiple systems and a lack of red blood cells.5 In erythroid cells, LDB1 forms a complex with DNA-binding proteins SCL/TAL1 and GATA1 and bridging protein LMO2, an erythroid LIM-only protein. The complex links globin gene promoters to the β-globin locus control region enhancer by LDB1 self-interaction and functions as the primary mediator of global erythroid gene activation.6,7
Prior to Schuetzmann et al, the role of LDB1 in myeloid cells had been unclear. We now learn that LDB1 is expressed in THP-1 cells and is upregulated during differentiation. Furthermore, LDB1 is recruited to several positions within the PU.1 SubTAD, including the gene promoter and URE. Inducible knockdown of PU.1 resulted in substantial loss of LDB1 in the locus, indicating that PU.1 is required for LDB1 recruitment, although the effect may be indirect since PU.1 and LDB1 were not found to interact. Importantly, dox-inducible knockdown of PU.1 or LDB1 revealed distinctive functions. After differentiation-induced PU.1/URE contact was established, reduction of LDB1 resulted in lost contact, whereas PU.1 reduction had no effect. Therefore, contacts are maintained by LDB1. The figure depicts a speculative model incorporating these results.
This report affects several different lines of research. First, the work reinforces autoregulation of the master regulator of myelopoiesis PU.1, and we learn that its long-range contacts to the URE enhancer correlate with its expression in normal and AML cells. To establish a causal role, it would be interesting to see if an LDB1 targeting approach, like that taken by Deng et al,8 could reconnect PU.1 to its enhancer in AML cells and lead to a more normal cellular phenotype. Second, we learn that LDB1 both establishes and maintains PU.1 gene contact to the URE enhancer. This result nicely expands the mechanistic role of LDB1 in enhancer looping from erythroid cells to target genes characteristic of the myeloid lineage.
How is LDB1 recruited to the PU.1 gene and the URE? LDB1 requires DNA-binding partners and a LIM-domain cofactor to form an active complex and has a distinct E box/GATA compound motif recognized by SCL and GATA1 in erythroid cells.7 What motif underlies its sites of occupancy in myeloid cells that result in targeting of myeloid-specific genes and enhancers? Proteomics would be a complementary approach to reveal the nature of specific differences between the erythroid and myeloid LDB1 complexes. The more we understand about how long-range enhancer contacts are engineered, the more we will be able to use the information for therapeutic interventions.
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal