

In this issue of Blood, 1 report that the retained nuclear localization of the transcription factor forkhead box class O1 (FOXO1) is a lymphoma-promoting, oncogenic event in the pathogenesis of Burkitt lymphoma (BL). Recurrent mutations in FOXO1 have been reported in ∼10% of diffuse large B-cell lymphoma (DLBCL) and BL cases,2 but to date the functional consequences of these mutations in the context of germinal center (GC) B-cell lymphomagenesis remained incompletely understood.
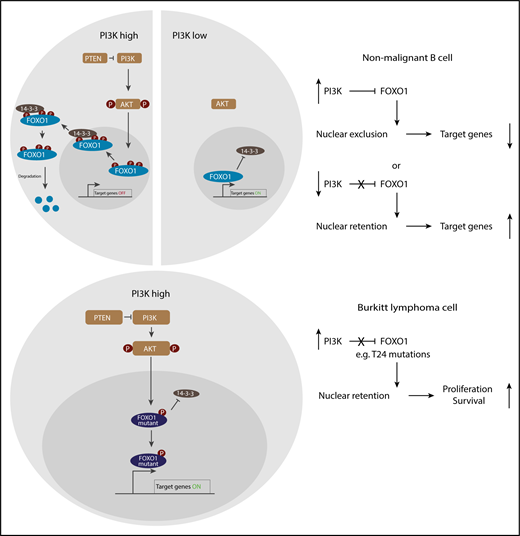
Activity of FOXO1 in nonmalignant and Burkitt lymphoma cells. High phosphoinositide-3-kinase (PI3K) kinase activity in nonmalignant cells leads to FOXO1 phosphorylation (P), followed by its 14-3-3 protein-associated nuclear export and proteasomal degradation, whereas FOXO1 regulates target genes in the absence of PI3K signaling. In Burkitt lymphoma cells, mutations in FOXO1 prevent its export from the nucleus in the presence of constitutive PI3K activity, promoting survival and proliferation.
Activity of FOXO1 in nonmalignant and Burkitt lymphoma cells. High phosphoinositide-3-kinase (PI3K) kinase activity in nonmalignant cells leads to FOXO1 phosphorylation (P), followed by its 14-3-3 protein-associated nuclear export and proteasomal degradation, whereas FOXO1 regulates target genes in the absence of PI3K signaling. In Burkitt lymphoma cells, mutations in FOXO1 prevent its export from the nucleus in the presence of constitutive PI3K activity, promoting survival and proliferation.
FOXO1 plays an important role in early B-cell development as well as in the differentiation of GC B cells in the dark zone.3,4 Several lines of evidence support a tumor suppressor role of FOXO1 in the development of hematopoietic malignancies, for example, in classical Hodgkin lymphoma.5 Mutations in FOXO1 in DLBCL however not only were reminiscent of oncogenic rather than tumor suppressor mutations (few hotspots mutations that cause amino acid exchanges rather than a broad distribution of destructive mutations) but also were also shown to maintain nuclear localization and possibly transcription factor activity.6
Phosphorylation by the Akt kinase critically controls FOXO1 localization, as 14-3-3 proteins bind to the phosphorylated site of FOXO1 and assist its nuclear export. FOXO1 contains 3 Akt recognition motifs (consensus sequence RXRXXS/T), of which the most N-terminal one around threonine 24 (T24) and the one in the forkhead box domain around serine 256 (S256) are crucial for FOXO1 nuclear export (see figure).7
PI3K/Akt signaling downstream of constitutive B-cell receptor signaling, which is at least in part due to mutations in TCF3/ID3, cooperates with high MYC expression due to chromosomal translocations to promote BL development.8,9 It is therefore expected that FOXO1 is continuously phosphorylated and exported from the nucleus. However, nuclear FOXO1 was detected in cells from human BL-derived cell lines and even in the lymphoma cells from a BL mouse model that is driven by constitutive MYC and P110 transgene expression.9 Kabrani et al then identified recurrent mutations in FOXO1 in human BL-derived cell lines and in murine BL-like cells (similar to the ones in DLBCL) that prevented phosphorylation by Akt including T24 amino acid exchanges. Consistent with an earlier study,6 these mutations caused the predominantly nuclear localization of mutated FOXO1 also in BL cells, yet FOXO1 was detected in the nucleus of cells of the human BL cell lines independent of its mutation status.
Murine BL-like cells with T24 mutation but not with wild-type FOXO1 status showed increased cell death and impaired cell-cycle progression after Cas9/gRNA–mediated FOXO1 deletion. Of note, murine BL-like cells with mutated FOXO1 were still dependent on PI3K signaling as in-frame mutations following CRISPR/Cas9–mediated knockout of the P110 transgene showed preferential selection over time.
Gene expression profiling analysis of murine BL-like cells after Cas9-editing of FOXO1 revealed a “dominant effect” of mutated FOXO1 (independent of wild-type allele) on a subset of genes: cell cycle, cell division, and proliferation ontologies were enriched in BL cells with mutated FOXO1 and genes lower expressed in these cells included regulators of apoptosis and metabolism. These seem obvious functional targets, but a contribution of other processes that FOXO1 is involved in, such as metabolism, autophagy, and oxidative stress,5 cannot be excluded. Mutant, nuclear FOXO1 likely maintains the expression of the target genes in BL cells that FOXO1 induces in dark zone GC B cells, suggesting that mutant and wild-type FOXO1 are transcriptionally and functionally similar. The T24 FOXO1 mutation did compromise S256 phosphorylation and 14-3-3 binding, but other posttranslation modifications (ie, lysine acetylation and O-glycosylation) were not different from those of wild-type FOXO1 in BL cells.
To determine whether nuclear, wild-type FOXO1 is sufficient to support lymphoma cell growth even in the presence of T24 phosphorylation, the authors created a FOXO1 mutant that eliminated the nuclear export signal, FOXO1-L375A. Expression of FOXO1-L375A in a FOXO1-knockout background of FOXO1-mutated murine BL cells (because they require nuclear FOXO1) was able to functionally compensate for T24-mutated FOXO1 and support lymphoma cell growth. Whether nuclear FOXO1 in primary human BL samples may be of clinical relevance and may serve as prognostic marker6 remains to be clarified.
The findings by Kabrani and colleagues shed light on the functional role of FOXO1 in BL pathogenesis. Oncogenic FOXO1 activity in the presence of constitutive PI3/Akt signaling was mediated at least in part due to mutations that cause nuclear retention of the transcription factor (see figure). At the same time, the mechanism(s) that grant coexistence of nuclear FOXO1 and PI3K/Akt activity in the absence of FOXO1 mutations require further investigation; certainly, other signaling pathways affect FOXO1 shuttling.5,7 With respect to the “2 faces” of FOXO1, it is tempting to speculate that similar to the situation in other cancers10 the spatial-temporal activity and optimal balance of FOXO1 tip the balance toward either tumor promotion or tumor suppression.
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal