

TO THE EDITOR:
Follicular lymphoma (FL) is characterized by the t(14;18)(q32;q21) chromosomal translocation,1 found in ∼85% of manifest FL (mFL) cases. The t(14;18) is also present in early precursor lesions of FL and in a significant fraction of healthy individuals.2,3 This translocation occurs early in B-cell development resulting from a repair error during the variable diversity joining recombination process.4 The t(14;18) alone is not sufficient to drive FL lymphomagenesis, indicating the need for additional genetic hits.5 Another feature of FL cells is the constitutive expression of activation-induced cytidine deaminase, which mediates the processes of somatic hypermutation (SHM) and class switch recombination, leading to genomic instability and accumulation of oncogenic aberrations.6 Accordingly, secondary hits are frequently identified in mFL, including chromosomal alterations and mutations targeting (eg, acetyl- and methyltransferase genes).7-10 Such events occur also in t(14;18)+ cells of healthy individuals and in in situ follicular neoplasia (ISFN).11-13
These findings suggest a possible model of FL lymphomagenesis in which the t(14;18) is the first hit leading to the accumulation of cells that will then undergo repetitive rounds of germinal center reaction, acquiring additional hits by SHM and class switch recombination, some of which might provide a selective advantage and drive malignant progression.14 But the particular events needed to transform the ISFN cells into mFL are largely unknown. A hierarchical model recently described CREBBP mutations as early events in FL evolution.15,16 In contrast, KMT2D and TNFRSF14 mutations are reported as late events that function as possible accelerator mutations.16
The aim of our study was to identify mutations in ISFN that might represent early driver mutations in the genomic evolution of FL. For this purpose, 5 ISFN-only cases (nonprogressing) and 6 clonally related samples of ISFN and mFL were subjected to analysis (Table 1). The paired ISFN/mFL cases have been the subject of previous studies.13,17 For this analysis, the ISFN cases were microdissected as previously reported.13,18 The diagnosis of ISFN and mFL was performed according to the 2016 revised World Health Organization classification.1 Four mFL cases (mFL-3 to 6) were analyzed by next-generation sequencing (NGS) using an Ion AmpliSeq Custom Panel covering all exons of TNFRSF14, KMT2D, FOXO1, EP300, MEF2B, HIST1H1B-E, and GNA13, as well as hot spot regions of EZH2 (exon 16) and CREBBP (exons 24-28 and 30). NGS analysis was done on the Ion Torrent PGM technology, as previously described.19 mFL cases 1 and 2 were Sanger sequenced for the 2 most recurrently mutated genes EZH2 (exon 16) and CREBBP (exons 26-28 and 30) only.19 To examine if the mutations found in the 6 mFL cases were present in the paired ISFN cases, we sequenced these regions in the ISFN cases using targeted resequencing and/or Sanger sequencing (supplemental Tables 1 and 2, available on the Blood Web site). We extended the analysis to 5 ISFN-only cases (ISFN-7 to 11) by NGS using the same AmpliSeq Custom Panel. Additionally, we performed NGS-based B-cell clonality analysis and determination of N-glycosylation sites (supplemental Materials and methods). The study was approved by the local ethics committees of participating institutions (UKT 219/2012/BO2).
Mutation overview in ISFN/mFL pairs and ISFN-only cases
| Case no. | Gene | Protein level | DNA level | Coverage | Frequency, % | Verification* | Method | ISFN no. | Analysis in paired ISFN |
|---|---|---|---|---|---|---|---|---|---|
| ISFN/mFL pairs | |||||||||
| mFL-1 | CREBBP | p.L1499Q | c.4496T>A | ND | ND | — | Sanger | 1 | Not present† |
| mFL-2 | EZH2 | p.Y646N | c.1936T>A | ND | ND | — | Sanger | 2 | Not present† |
| CREBBP | p.I1471T | c.4412T>C | ND | ND | — | Present‡ | |||
| mFL-3 | TNFRSF14 | p.C185S | c.553T>A | 135 | 62.2 | Confirmed | NGS | 3 | Present (9%)† |
| CREBBP | p.L1434P | c.4301T>C | 2541 | 29.7 | Confirmed | Present (3%)† | |||
| CREBBP | p.R1446H | c.4337G>A | 2515 | 29.2 | Confirmed | Present (6%)†,‡ | |||
| EZH2 | p.Y646H | c.1936T>C | 1190 | 30.4 | Confirmed | Not present but p.Y646N (5%)† | |||
| KMTD2 | p.K1840fs | c.5519_5529delAAGCCGATACA | 102 | 43.6 | Confirmed | Present‡ | |||
| mFL-4 | TNFRSF14 | p.S171C | c.512C>G | 777 | 59.6 | Confirmed | NGS | 4 | Not present†,‡ |
| EZH2 | p.Y646N | c.1936T>A | 1482 | 48.6 | Confirmed | Present (33%)† | |||
| CREBBP | p.L1499P | c.4496T>C | 3022 | 74.8 | Confirmed | Present (20%)† | |||
| mFL-5 | CREBBP | p.S1382fs | c.4145delA | 1663 | 20.4 | Confirmed | NGS | 5 | Present (15%)† |
| EP300 | p.D1399Y | c.4195G>T | 532 | 17.5 | Confirmed | Not present† | |||
| mFL-6 | CREBBP | p.R1446C | c.4336C>T | 1021 | 6.2 | Confirmed | NGS | 6 | Present (5%)† |
| EZH2 | p.Y646F | c.1937A>T | 498 | 5.0 | Confirmed | Present†,‡ | |||
| ISFN-only cases | |||||||||
| ISFN-7 | KMT2D | p.C5227Ter | c.15681C>A | 6012 | 14 | Confirmed | NGS | ||
| KMT2D | p.Q3518Ter | c.10552C>T | 1064 | 15 | Confirmed | — | — | ||
| ISFN-8 | EP300 | p.T1332P | c.3994A>C | 4651 | 11 | Confirmed | NGS | ||
| TNFRSF14 | Splice site | c.179-2A>T | 9554 | 10 | Confirmed | — | — | ||
| ISFN-9 | TNFRSF14 | p.C121Ter | c.363C>A | 6251 | 8 | Confirmed | NGS | ||
| CREBBP | p.H1487Y | c.4459C>T | 6562 | 12 | Confirmed | — | — | ||
| ISFN-10 | EZH2 | p.Y646F | c.1937A>T | 789 | 3 | Confirmed | NGS | — | — |
| ISFN-11 | — | — | — | — | — | — | NGS | — | — |
| Case no. | Gene | Protein level | DNA level | Coverage | Frequency, % | Verification* | Method | ISFN no. | Analysis in paired ISFN |
|---|---|---|---|---|---|---|---|---|---|
| ISFN/mFL pairs | |||||||||
| mFL-1 | CREBBP | p.L1499Q | c.4496T>A | ND | ND | — | Sanger | 1 | Not present† |
| mFL-2 | EZH2 | p.Y646N | c.1936T>A | ND | ND | — | Sanger | 2 | Not present† |
| CREBBP | p.I1471T | c.4412T>C | ND | ND | — | Present‡ | |||
| mFL-3 | TNFRSF14 | p.C185S | c.553T>A | 135 | 62.2 | Confirmed | NGS | 3 | Present (9%)† |
| CREBBP | p.L1434P | c.4301T>C | 2541 | 29.7 | Confirmed | Present (3%)† | |||
| CREBBP | p.R1446H | c.4337G>A | 2515 | 29.2 | Confirmed | Present (6%)†,‡ | |||
| EZH2 | p.Y646H | c.1936T>C | 1190 | 30.4 | Confirmed | Not present but p.Y646N (5%)† | |||
| KMTD2 | p.K1840fs | c.5519_5529delAAGCCGATACA | 102 | 43.6 | Confirmed | Present‡ | |||
| mFL-4 | TNFRSF14 | p.S171C | c.512C>G | 777 | 59.6 | Confirmed | NGS | 4 | Not present†,‡ |
| EZH2 | p.Y646N | c.1936T>A | 1482 | 48.6 | Confirmed | Present (33%)† | |||
| CREBBP | p.L1499P | c.4496T>C | 3022 | 74.8 | Confirmed | Present (20%)† | |||
| mFL-5 | CREBBP | p.S1382fs | c.4145delA | 1663 | 20.4 | Confirmed | NGS | 5 | Present (15%)† |
| EP300 | p.D1399Y | c.4195G>T | 532 | 17.5 | Confirmed | Not present† | |||
| mFL-6 | CREBBP | p.R1446C | c.4336C>T | 1021 | 6.2 | Confirmed | NGS | 6 | Present (5%)† |
| EZH2 | p.Y646F | c.1937A>T | 498 | 5.0 | Confirmed | Present†,‡ | |||
| ISFN-only cases | |||||||||
| ISFN-7 | KMT2D | p.C5227Ter | c.15681C>A | 6012 | 14 | Confirmed | NGS | ||
| KMT2D | p.Q3518Ter | c.10552C>T | 1064 | 15 | Confirmed | — | — | ||
| ISFN-8 | EP300 | p.T1332P | c.3994A>C | 4651 | 11 | Confirmed | NGS | ||
| TNFRSF14 | Splice site | c.179-2A>T | 9554 | 10 | Confirmed | — | — | ||
| ISFN-9 | TNFRSF14 | p.C121Ter | c.363C>A | 6251 | 8 | Confirmed | NGS | ||
| CREBBP | p.H1487Y | c.4459C>T | 6562 | 12 | Confirmed | — | — | ||
| ISFN-10 | EZH2 | p.Y646F | c.1937A>T | 789 | 3 | Confirmed | NGS | — | — |
| ISFN-11 | — | — | — | — | — | — | NGS | — | — |
5% VAF was defined as sensitivity threshold in the analysis of manifest FL samples.
ND, no data available.
Verification of mutations in mFL was performed either with Sanger sequencing or Illumina MiSeq; verification of mutations in ISFN-only cases was performed with single amplicons on the Ion Torrent PGM.
Analysis by targeted resequencing on the Illumina MiSeq.
Analysis with Sanger sequencing.
The clinical data and genetic analyses are summarized in supplemental Tables 3 and 4. In 4 informative ISFN cases, recurrent N-glycosylation motifs (N-X-S/T) were identified in the CDR3 and CDR2/FR2 regions. This finding indicates that, already at this early stage, N-glycosylation sites occur and substitute conventional antigen binding favoring the generation of long-lived ISFN clones and increasing the chance of secondary hits that will further drive clone fate.20,21 Sequencing analysis of the 6 mFL cases revealed CREBBP as the most frequently mutated gene (6 cases; 100%) (mFL-3 with 2 mutations). CREBBP was followed by mutations in EZH2 (4 cases), TNFRSF14 (2 cases), EP300 (1 case), and KMT2D (1 case) (Figure 1A). The allelic frequencies of these mutations ranged from 5% to 74.8% (mean, 35.6%) (Table 1). The highest mutant allelic frequencies were detected in CREBBP (mFL-4, 74.8%) and TNFRSF14 (mFL-3, 62.2%; mFL-4, 59.6%), suggesting additional loss or copy number neutral loss of heterozygosity of 16p13 and 1p36 chromosomal regions. Accordingly, comparative genomic hybridization array analysis showed loss of 1p36 region in case mFL-4 (supplemental Figure 1).13
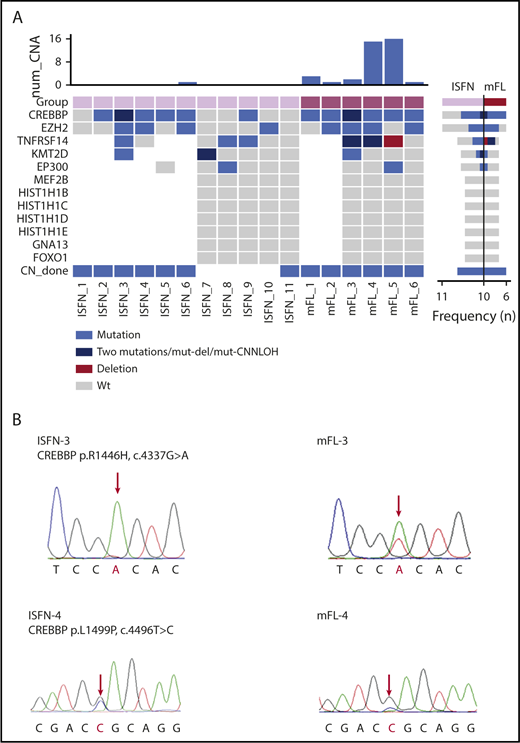
Mutation overview in mFL and ISFN cases. (A) The heat map shows the case-specific pattern of mutations found by Sanger sequencing and NGS. Each column represents a case sorted by ISFN and mFL. Each row represents a gene. (Right) Bar graph illustrates the mutation frequency of each gene in a specific entity. (Top) Bar graph indicates the number of copy number alterations detected by array-comparative genomic hybridization in a previous study.13 Loss of heterozygosity determination based on polymorphic single-nucleotide polymorphism (data not shown) indicated TNFRSF14 CNN-LOH alteration in mFL-3. mFL cases 1 and 2 were analyzed for mutations in CREBBP and EZH2 with Sanger sequencing only. (B) Examples of CREBBP mutations detected in 2 paired ISFN/mFL samples by Sanger sequencing. The peak of the ancestral base is higher than in the mFL case in the ISFN. The analysis was performed in microdissected ISFN. CNN-LOH, copy number neutral loss of heterozygosity; wt, wild-type.
Mutation overview in mFL and ISFN cases. (A) The heat map shows the case-specific pattern of mutations found by Sanger sequencing and NGS. Each column represents a case sorted by ISFN and mFL. Each row represents a gene. (Right) Bar graph illustrates the mutation frequency of each gene in a specific entity. (Top) Bar graph indicates the number of copy number alterations detected by array-comparative genomic hybridization in a previous study.13 Loss of heterozygosity determination based on polymorphic single-nucleotide polymorphism (data not shown) indicated TNFRSF14 CNN-LOH alteration in mFL-3. mFL cases 1 and 2 were analyzed for mutations in CREBBP and EZH2 with Sanger sequencing only. (B) Examples of CREBBP mutations detected in 2 paired ISFN/mFL samples by Sanger sequencing. The peak of the ancestral base is higher than in the mFL case in the ISFN. The analysis was performed in microdissected ISFN. CNN-LOH, copy number neutral loss of heterozygosity; wt, wild-type.
In the 6 clonally related ISFN cases, surprisingly, 10 of 15 mutations found in mFL were already present in ISFN, including 6/7 CREBBP mutations, 2/4 EZH2 mutations, 1/2 TNFRSF14 mutations, and 1/1 KMT2D mutation. Only 1 ISFN case (ISFN-1) showed no mutations, despite the presence of a CREBBP mutation in the corresponding mFL. Comparison of the allelic frequencies of the mutations in mFL and microdissected ISFN revealed lower frequencies in the ISFN cases (mean, 10.4% vs 35.6%). Sanger sequencing analysis also suggested a lower CREBBP mutation allelic frequency in ISFN cases (Figure 1B). Interestingly, 1 ISFN case carried mutations in CREBBP, TNFRSF14, and KMT2D identical to the corresponding mFL, but a different EZH2 mutation (ISFN-3). This confirms the existence of divergent clonal evolution early in disease progression of FL.13
ISFN cases without evidence of mFL (ISFN-7 to 11) carried mutations in 4/5 cases. Mutations were detected in KMT2D (case ISFN-7 with 2 mutations), TNFRSF14 (2 cases), CREBBP (1 case), EZH2 (1 case), and EP300 (1 case). The allelic frequencies ranged from 8% to 15% (mean, 11.7%) in microdissected cases. Analyses by sorting intolerant from tolerant, PolyPhen 2, and combined annotation-dependent depletion predicted a damaging effect for all mutations (supplemental Table 5).
Although this is a small cohort and results should be taken with caution, the frequent occurrence of CREBBP mutations in 5/6 paired ISFN cases, but in only 1/5 ISFN-only cases (83% vs 20%) suggests that this mutation might be needed for the transforming event. Mutations in CREBBP are inactivating events that impair the acetylation activity of CREBBP, favoring the constitutive activity of BCL6 and decreased p53 activity. Recently, it has been shown that loss of CREBBP abrogates optimal cellular response to DNA damage because of defective p53 activity, creating a permissive state for the retention and accumulation of subsequent mutations, facilitating transformation.10,22 Furthermore, FL with CREBBP mutations showed significantly higher aberrant SHM-induced immunoglobulin heavy chain variable divergence, highlighting CREBBP as a key protein generating genetic and epigenetic coevolution.15 Accordingly, our data suggest that mutations in CREBBP represent early driver mutations probably facilitating the acquisition of additional mutations and improving the “clonal fitness” of the cells.15 Whether CREBBP mutations alone are sufficient for transformation is not clear; however, a recent study suggests that earlier loss of CREBBP is advantageous for lymphoid transformation and final evolution of lymphoid malignancies.22
EZH2 Tyr646 mutation was the second most common aberration found in ISFN, indicating that it might represent an early event.16,23,24 Mutations in KMT2D and TNFRSF14 have been shown to be late events in FL evolution.16 However, we now show that these mutations also occur in ISFN. Accordingly, a recent mutational analysis on duodenal-type FL, another FL variant considered an early precursor lesion localized to the intestine, demonstrated frequent mutations in CREBBP followed by mutations in EZH2 and TNFRSF14 genes.25 However, KMT2D was less commonly mutated than in mFL, further supporting that KMT2D mutations are late events in FL pathogenesis.
In conclusion, this study shows for the first time that mutations in CREBBP, EZH2, TNFRSF14, and KMT2D are already present in ISFN but with different frequencies.18 The possible role of CREBBP mutations in the transformation of a subclone into overt mFL warrants further analysis.
The online version of this article contains a data supplement.
Acknowledgments
This work was supported by grants from the Deutsche Forschungsgemainschaft (QU144/1-1 [L.Q.-M.], FE597/4-1 [F.F.], and QUI44/1-1 [I.M.]), and a fellowship from Generalitat de Catalunya AGAUR FI-DGR 2017 (2017 FI_B01004) (J.E.R.-Z.).
Authorship
Contribution: L.Q.-M. conceived and designed the study, supervised the experimental work, and wrote the manuscript; J. Schmidt performed genetic analysis, interpreted the data, and helped write the manuscript; J.E.R.-Z., I.S., I.B., and R.S. interpreted the data and helped write the manuscript; J. Steinhilber, A.H., I.M., S.C.Y., and M.R. performed experimental work; E.S.J., F.F., and L.Q.-M. provided and reviewed the cases; and E.S.J. and F.F. helped write the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Leticia Quintanilla-Martinez, Institute of Pathology, University Hospital Tübingen, Eberhard-Karls-University of Tübingen and Comprehensive Cancer Center, Liebermeisterstr 8, 72076 Tübingen, Germany; e-mail: leticia.quintanilla-fend@med.uni-tuebingen.de.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal