

Key Points
Donor T cells lacking Nrf2 demonstrate ameliorated GVHD.
Absence of Nrf2 on donor cells enhanced the persistence of Helios+ natural regulatory T cells in allograft recipients.

Abstract
Nuclear factor erythroid-derived 2-like 2 (Nrf2) is a ubiquitously expressed transcription factor that is well known for its role in regulating the cellular redox pathway. Although there is mounting evidence suggesting a critical role for Nrf2 in hematopoietic stem cells and innate leukocytes, little is known about its involvement in T-cell biology. In this study, we identified a novel role for Nrf2 in regulating alloreactive T-cell function during allogeneic hematopoietic cell transplantation (allo-HCT). We observed increased expression and nuclear translocation of Nrf2 upon T-cell activation in vitro, especially in CD4+ donor T cells after allo-HCT. Allo-HCT recipients of Nrf2−/− donor T cells had significantly less acute graft-versus-host disease (GVHD)-induced mortality, morbidity, and pathology. This reduction in GVHD was associated with the persistence of Helios+ donor regulatory T cells in the allograft, as well as defective upregulation of the gut-homing receptor LPAM-1 on alloreactive CD8+ T cells. Additionally, Nrf2−/− donor CD8+ T cells demonstrated intact cytotoxicity against allogeneic target cells. Tumor-bearing allo-HCT recipients of Nrf2−/− donor T cells had overall improved survival as a result of preserved graft-versus-tumor activity and reduced GVHD activity. Our findings characterized a previously unrecognized role for Nrf2 in T-cell function, as well as revealed a novel therapeutic target to improve the outcomes of allo-HCT.
Introduction
Allogeneic hematopoietic cell transplantation (allo-HCT) is an important therapy with curative potential for patients with hematologic malignancies. The therapeutic benefits of allo-HCT are derived from high doses of cytoreductive conditioning and the immune-mediated graft-versus-tumor (GVT) effect. However, the deleterious side effect to the beneficial GVT activity is acute graft-versus-host disease (GVHD). GVHD is a systemic inflammatory disease that affects 40% to 60% of allo-HCT patients and accounts for 15% of deaths after allo-HCT,1 thereby limiting the success and wider application of allo-HCT, despite its curative potential. Although a variety of immune and nonimmune cells are involved, allogeneic donor T lymphocytes are the primary effectors and regulators of GVT and GVHD responses.2 Therefore, separation of the undesired GVHD activities and beneficial GVT activities of alloreactive T (allo-T) cells remains critical for the improvement of clinical outcomes after allo-HCT.
Nuclear factor erythroid-derived 2-like 2 (NFE2L2, or Nrf2) is a ubiquitously expressed basic leucine zipper transcription factor that can function as a master regulator of cellular redox, detoxification, and cellular stress pathways.3-5 The dual roles of Nrf2 in cancer promotion and cancer prevention in various solid tumors have been widely studied and have demonstrated importance in tumorigenesis.6,7 Moreover, we have recently shown that Nrf2 positively regulates the self-renewal ability of hematopoietic stem cells.8 In addition, Nrf2 expression has been implicated in the resistance of leukemia and lymphoma cells to apoptosis.9-11 Interestingly, recent reports suggest that genetic Nrf2 activation has an anti-inflammatory effect in an ischemia-reperfusion–induced acute kidney injury model and in Scurfy mice.12,13 Given that inhibition of the Nrf2 pathway could represent an attractive therapeutic approach for hematologic malignancies, we investigated the consequences of Nrf2 inhibition in allo-T cells in an effort to develop adjuvant therapies to mitigate GVHD and maintain tumor clearance in the context of allo-HCT. We hypothesized that Nrf2 is involved in T-cell alloreactivity, and we sought to analyze how Nrf2 disruption in donor T cells affects their ability to cause GVHD and GVT using genetically altered Nrf2−/− mice.
Methods
Mice
Nrf2−/− mice, a kind gift from Jefferson Chan (University of California, Irvine, CA), were backcrossed onto the C57BL/6 (B6) (CD45.2 B6, H-2b) background for ≥10 generations. Wild-type (WT; Nrf2+/+) B6 (CD45.2 B6, H-2b), B6.SJL-PtprcaPepcb/BoyJ (CD45.1 B6 congenic, H-2Kb), BALB/c (H-2d), and LP (H-2Kb) mice were purchased from The Jackson Laboratory (Bar Harbor, ME). All mice were female Nrf2+/+ or Nrf2−/−, 8 to 12 weeks old, and maintained in the Memorial Sloan Kettering Cancer Center (MSKCC) Animal Facility in Thorensten units with filtered germ-free air. Experiments were conducted in compliance with the institutional guidelines at MSKCC.
Cell isolation
Bone marrow (BM) cells were flushed from intact femurs and tibiae, and spleens were mashed with glass slides to generate single-cell suspensions. The cells were collected in RPMI 1640 media containing 10% fetal bovine serum or phosphate-buffered saline and 0.5% bovine serum albumin and filtered through a 70-μm strainer. T-cell–depleted (TCD) BM cells were prepared by incubation with anti–Thy-1.2 and low–TOX-M rabbit complement (Cedarlane Laboratories, Hornby, ON, Canada). CD5+, CD4+, CD8+, and CD4+CD25+ regulatory T cells (Tregs) and naive CD4+ T cells were enriched from splenocytes using Miltenyi Biotec MACS purification kits (Auburn, CA), as indicated.
Hematopoietic cell transplantation
The hematopoietic cell transplantation (HCT) procedure was performed as previously described.14 Briefly, CD45.1 B6 congenic TCD BM cells (5 × 106) were administered via tail vein injection into lethally irradiated hosts (850 cGy, split-dose for BALB/c recipients or 1100 cGy, split-dose for CD45.1 congenic B6 or LP recipients). Unless otherwise specified, CD5-enriched T cells from CD45.2 B6 (WT or Nrf2−/−) donors were transplanted into BALB/c (0.5 × 106) or LP allogeneic (2 × 106) recipients. Where indicated, a total of 0.5 × 106 splenic B6 CD4+ and CD8+ T cells, mixed at a physiological ratio (2:1),15 was transplanted into BALB/c hosts. Recipient mice were monitored for survival and clinical GVHD symptoms or were euthanized for blinded histopathologic and flow cytometric analysis, as previously described.16,17 In GVT experiments, BALB/c recipients received 0.25 × 106 A20 B-cell lymphoma cells (BALB/c background) on the day of transplantation in addition to donor BM, with or without donor T cells. Mice were monitored for survival and tumor-related morbidity, and the cause of death was determined by autopsy or histopathology, as previously described.16 In in vivo cytotoxicity assays, a total of 20 × 106 carboxyfluorescein succinimidyl ester (CFSE)-labeled (Thermo Fisher Scientific, Waltham, MA) BALB/c allogeneic target splenocytes was mixed with PKH26-labeled (Sigma-Aldrich, St. Louis, MO) syngeneic control B6 target splenocytes at a 1:1 ratio, injected on day 7 after allo-HCT, and analyzed 4 hours later.
Flow cytometry
All antibodies were obtained from BD Pharmingen (San Jose, CA), with the exception of those against Nrf2 (Abcam, Burlingame, CA); CD44 (IM7), T-bet (4B10), Helios (22F6) (BioLegend, San Diego, CA); FoxP3 (FJK-16S), H-2Kb (AF6-88.5), H-2Kd (SF1-1.1) CCR9 (CW-1.2), RORγt (AFKJS) (eBioscience, San Diego, CA); and neuropilin-1 (3DS304M) (R&D Systems, Minneapolis, MN). Surface staining was performed for 15 minutes using the indicated mixture of antibodies, whereas intracellular staining was performed using a Fixation/Permeabilization kit (eBioscience, San Diego, CA), according to the manufacturer’s protocol.
Immunofluorescence
T cells (0.5 × 106) were spun into each well of 4-well Millicell glass chamber slides (EMD Millipore, Burlington, MA) by low-speed centrifugation (300 rpm) for 3 minutes and then fixed with 4% paraformaldehyde (Sigma-Aldrich) for 20 minutes. After washing with PBS, cells were permeabilized in 0.1% Triton™ X-100 and incubated in 1% bovine serum albumin overnight with polyclonal anti-Nrf2 antibody (1:250; Abcam, Burlingame, CA). Cells were washed and stained with Alexa Fluor 488 goat anti-rabbit IgG antibody (1:500; Invitrogen, Waltham, MA), and nuclei were stained with 5 μg/mL 4′,6-diamidino-2-phenylindole, dihydrochloride (DAPI; Invitrogen, Waltham, MA). Images were captured on an LSM 880 confocal microscope (Zeiss, Oberkochen, Germany). Scanned images were analyzed using Fiji software as reported.18 Briefly, cell segmentation using the DAPI channel was smoothed over using a median filter and then thresholded to create a mask to obtain round cells. Overlapping cells were removed by dilating the DAPI mask using the same size and circularity filter. The DAPI mask was used to create the cell and cytoplasmic masks by dilation, erosion, and image subtraction. The signal in the other channels can be separated out and averaged to create the intensities in each part of the cell using the masks.
Bioluminescence imaging
Animals that received A20-TGL19 were given 150 mg/kg D-Luciferin (Xenogen, Alameda, CA) intraperitoneally. The tumor burden for an individual mouse was determined by the total flux (photons per second) of the whole-body bioluminescent signal, which was superimposed on the conventional grayscale photographs, as previously described.16
Statistical analysis
Data were processed in GraphPad Prism 5.0 software. Statistical comparisons between 2 groups were performed with the nonparametric unpaired Mann–Whitney U test. Survival data were analyzed with the Mantel–Cox log-rank test. P values < .05 were considered statistically significant. All data shown in graphs represent the mean ± standard error of the mean (SEM) of each group.
Results
T-cell activation promotes Nrf2 nuclear translocation and protein expression
To define the role of Nrf2 in T-cell alloreactivity after allo-HCT, we first assessed the expression of Nrf2 in activated T cells. We found that total cellular, as well as nuclear, Nrf2 expression in T cells was significantly increased 24 hours after T-cell receptor (TCR) stimulation in vitro with anti-CD3 and anti-CD28 (Figure 1A-D). We next examined how Nrf2 deficiency affects T-cell alloreactivity in a well-established major histocompatibility complex (MHC)-disparate murine allo-HCT model (B6 → BALB/c). Compared with syngeneic controls, allogeneic donor–derived T cells, specifically the CD4+ subset, significantly upregulated intracellular Nrf2 (Figure 1E-F). Taken together, these findings suggest a role for Nrf2 in T-cell (allo)activation in vitro and in vivo.
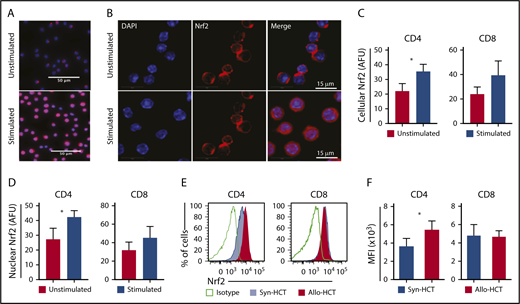
T cell-activation promotes Nrf2 nuclear translocation and protein expression. (A-D) Magnetically sorted WT B6 CD4 or CD8 T cells were stimulated with anti-CD3 and anti-CD28 for 24 hours for immunofluorescence analysis (n = 3 independent experiments). (A-B) Representative immunofluorescence images showing Nrf2 immunostaining (red) and DAPI (blue). Mean total cellular (C) and nuclear (D) fluorescent intensity of Nrf2 immunostaining. More than 600 cells were analyzed for each condition. (E-F) WT B6 TCD BM and T cells were transplanted into lethally irradiated B6 (Syn-HCT) or BALB/c (Allo-HCT) recipients. Representative flow cytometric analysis (E) and mean fluorescence intensity (MFI) (F) of the intracellular levels of Nrf2 within donor T cells were performed using recipient spleens on day 14 after HCT. Data represent the mean + standard deviation from 1 of 2 independent experiments (n = 5-7 mice per group). *P < .05. AFU, arbitrary fluorescent unit.
T cell-activation promotes Nrf2 nuclear translocation and protein expression. (A-D) Magnetically sorted WT B6 CD4 or CD8 T cells were stimulated with anti-CD3 and anti-CD28 for 24 hours for immunofluorescence analysis (n = 3 independent experiments). (A-B) Representative immunofluorescence images showing Nrf2 immunostaining (red) and DAPI (blue). Mean total cellular (C) and nuclear (D) fluorescent intensity of Nrf2 immunostaining. More than 600 cells were analyzed for each condition. (E-F) WT B6 TCD BM and T cells were transplanted into lethally irradiated B6 (Syn-HCT) or BALB/c (Allo-HCT) recipients. Representative flow cytometric analysis (E) and mean fluorescence intensity (MFI) (F) of the intracellular levels of Nrf2 within donor T cells were performed using recipient spleens on day 14 after HCT. Data represent the mean + standard deviation from 1 of 2 independent experiments (n = 5-7 mice per group). *P < .05. AFU, arbitrary fluorescent unit.
Nrf2−/− donor T cells induce less GVHD mortality and morbidity
To determine whether Nrf2 contributes to GVHD activity, we isolated Nrf2−/− splenic donor T cells and examined their ability to mediate acute GVHD. Importantly, spleens from WT and Nrf2−/− B6 donors had similar overall cellularity, T-cell numbers, and CD4+ and CD8+ TCR repertoire diversity at baseline (supplemental Figure 1A-B, available on the Blood Web site). We found that allo-HCT recipients of Nrf2−/− donor T cells had significantly lower GVHD mortality than did recipients of WT donor T cells (Figure 2A). Additionally, Nrf2−/− donor T cells mediated significantly less GVHD morbidity than WT cells, as measured by clinical GVHD scores and weight loss (Figure 2B-C). We found similar results in a second minor histocompatibility antigen–disparate model (B6 → LP) (Figure 2D-F). Taken together, these results in 2 GVHD models indicate a critical role for Nrf2 in GVHD-associated T-cell alloreactivity.
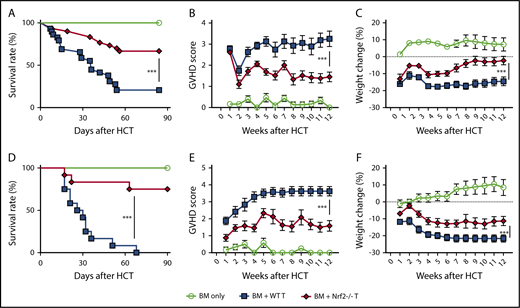
Nrf2−/−donor T cells induced less GVHD mortality and morbidity. (A-C) Lethally irradiated BALB/c recipients that received WT B6 TCD BM alone or with 0.5 × 106 B6 WT or Nrf2−/− T cells were monitored daily for Kaplan-Meier survival analysis (A) and weekly for GVHD clinical scores (B) and weight loss (C). Data represent the mean ± standard error of the mean from 3 independent experiments (BM, n = 14; BM + WT T, n = 29; BM + Nrf2−/− T, n = 30). Lethally irradiated LP recipients that received WT B6 TCD BM alone or with 2 × 106 B6 WT or Nrf2−/− T cells were monitored daily for survival (D) and weekly for GVHD clinical scores (E) and weight loss (F). Data represent the mean ± standard error of the mean from 3 independent experiments (n = 6 in BM group, n = 12 each in BM WT T and Nrf2−/− T groups). ***P < .005.
Nrf2−/−donor T cells induced less GVHD mortality and morbidity. (A-C) Lethally irradiated BALB/c recipients that received WT B6 TCD BM alone or with 0.5 × 106 B6 WT or Nrf2−/− T cells were monitored daily for Kaplan-Meier survival analysis (A) and weekly for GVHD clinical scores (B) and weight loss (C). Data represent the mean ± standard error of the mean from 3 independent experiments (BM, n = 14; BM + WT T, n = 29; BM + Nrf2−/− T, n = 30). Lethally irradiated LP recipients that received WT B6 TCD BM alone or with 2 × 106 B6 WT or Nrf2−/− T cells were monitored daily for survival (D) and weekly for GVHD clinical scores (E) and weight loss (F). Data represent the mean ± standard error of the mean from 3 independent experiments (n = 6 in BM group, n = 12 each in BM WT T and Nrf2−/− T groups). ***P < .005.
Nrf2 activity in allo-T cells contributes to hepatic, intestinal, and thymic GVHD
To determine whether the improved clinical outcomes in recipients of donor Nrf2−/− T cells could be attributed to reduced damage to GVHD target organs, we performed a semiquantitative histopathologic analysis. We found that recipients of Nrf2−/− T cells had significantly lower composite GVHD pathology scores in the liver, small bowel, and large bowel compared with recipients of WT T cells (Figure 3A-B). However, Nrf2 deficiency in donor T cells did not significantly impact the development of cutaneous GVHD, as indicated by the equivalent numbers of apoptotic keratinocytes in the epidermises of WT and Nrf2−/− T-cell recipients (Figure 3C). Additionally, donor allo-T cells mediate thymic GVHD, which leads to profound T-cell deficiency and a restricted TCR repertoire following allo-HCT.20-23 We found significantly less thymic GVHD in recipients of donor Nrf2−/− T cells, as measured by increases in overall thymic cellularity and the percentage of CD4+CD8+ double-positive thymocytes (Figure 3D-E). In conclusion, these findings suggest that Nrf2 activity in donor T cells contributes to the development of hepatic, gastrointestinal tract, and thymic, but not cutaneous, GVHD.
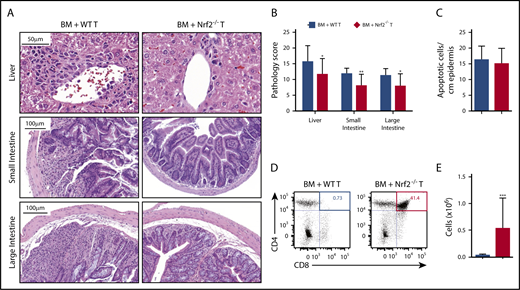
Nrf2 activity in allo-T cells contributes to hepatic, intestinal, and thymic GVHD. Lethally irradiated BALB/c recipients received B6 WT TCD BM and 0.5 × 106 B6 WT or Nrf2−/− T cells. The GVHD target organs of the recipients were analyzed on day 14 after HCT. Representative hematoxylin and eosin (H&E)-stained images of liver, small intestine, and large intestine (A) that were scored for histopathologic damage (B). (C) H&E-stained slides of skin were assessed for the number of apoptotic keratinocytes per centimeter of epidermis. Data represent the mean ± standard error of the mean combined from 2 independent experiments (n = 16 per group). Flow cytometric analysis of thymic damage; representative dot plots show the percentage (D) and number (E) of total CD4+CD8+ double-positive thymocytes. Data represent the mean + standard deviation combined from 3 independent experiments (n = 18 per group). *P < .05, **P < .01, ***P < .005.
Nrf2 activity in allo-T cells contributes to hepatic, intestinal, and thymic GVHD. Lethally irradiated BALB/c recipients received B6 WT TCD BM and 0.5 × 106 B6 WT or Nrf2−/− T cells. The GVHD target organs of the recipients were analyzed on day 14 after HCT. Representative hematoxylin and eosin (H&E)-stained images of liver, small intestine, and large intestine (A) that were scored for histopathologic damage (B). (C) H&E-stained slides of skin were assessed for the number of apoptotic keratinocytes per centimeter of epidermis. Data represent the mean ± standard error of the mean combined from 2 independent experiments (n = 16 per group). Flow cytometric analysis of thymic damage; representative dot plots show the percentage (D) and number (E) of total CD4+CD8+ double-positive thymocytes. Data represent the mean + standard deviation combined from 3 independent experiments (n = 18 per group). *P < .05, **P < .01, ***P < .005.
Nrf2−/− donor T cells display intact proliferation, activation, and apoptosis during the early phase of alloreactivity
Before infiltrating the target organs of recipients, and as early as 12 hours following transfer, donor T cells traffic to secondary lymphoid tissues where they undergo activation and start proliferating upon exposure to alloantigen.24-27 To assess whether the diminished GVHD activity of Nrf2−/− allo-T cells resulted from defective early alloactivation, we transferred CFSE-labeled donor T cells into MHC-disparate recipients and measured their proliferation and division indices (Figure 4A). Donor Nrf2−/− T cells displayed a modest and transient proliferation defect in alloantigen-responding CD4+ T cells, as well as in CD8+ T cells regardless of their antigen-encounter experience, during early alloactivation (Figure 4B).28 However, these defects appeared to be transient, as evidenced by the comparable proliferation and division indices, as well as the equivalent total numbers of highly proliferated (CFSElow) WT and Nrf2−/− donor CD4+ and CD8+ T cells at 96 hours after transplant (Figure 4B-C). Donor Nrf2−/− T cells also showed comparable activation, as measured by upregulation of CD25 and CD44, as well as downregulation of CD62L on the divided population (Figure 4D-E). Activated T cells may undergo activation-induced cell death, a physiologically important process to control clonal expansion of activated T cells and regulate immune responses.29,30 Donor Nrf2−/− T cells had a similar fraction of apoptotic cells compared with donor WT T cells during alloactivation, as measured by annexin V expression in the activated fractions (Figure 4F). These data suggest that donor Nrf2−/− T cells have a transient early proliferation defect that can contribute to, but is unlikely the main cause of, the striking clinical differences that we observed in our GVHD models.
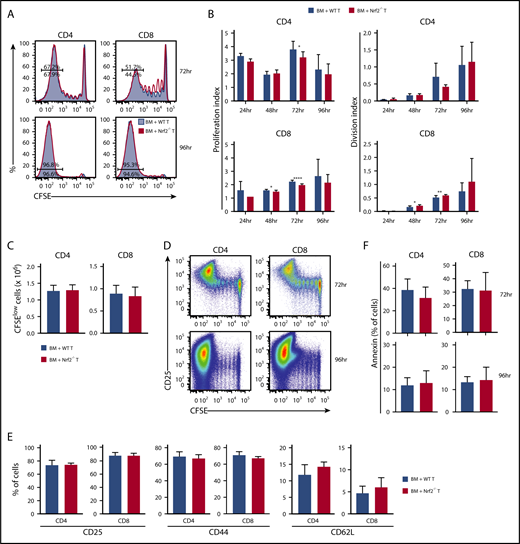
Nrf2−/−donor T cells display intact proliferation, activation, and apoptosis during the early phase of alloreactivity. Lethally irradiated BALB/c recipients received 5 × 106 CFSE-labeled B6 WT or Nrf2−/− T cells. Recipient spleens were analyzed by flow cytometry for CFSE dilution at the indicated time points. (A) Concatenated CFSE line graphs showing the percentages of CFSElow WT and Nrf2−/− donor T cells, gated on H-2Kb+ events. (B) Proliferation and division indices of WT and Nrf2−/− donor T cells. (C) Total numbers of CFSElow WT and Nrf2−/− donor T cells. (D) Representative flow cytometric analysis showing upregulation of the activation marker CD25 in the CFSElow population on WT donor T cells. Expression of activation markers at 96 hours (E) and apoptotic marker (F) on WT and Nrf2−/− donor T cells, gated on H2-Kb+CFSElow populations. Data represent the mean + standard deviation combined from 2 or 3 independent experiments (n ≥ 10 per group). *P < .05, **P < .01, ****P < .0001.
Nrf2−/−donor T cells display intact proliferation, activation, and apoptosis during the early phase of alloreactivity. Lethally irradiated BALB/c recipients received 5 × 106 CFSE-labeled B6 WT or Nrf2−/− T cells. Recipient spleens were analyzed by flow cytometry for CFSE dilution at the indicated time points. (A) Concatenated CFSE line graphs showing the percentages of CFSElow WT and Nrf2−/− donor T cells, gated on H-2Kb+ events. (B) Proliferation and division indices of WT and Nrf2−/− donor T cells. (C) Total numbers of CFSElow WT and Nrf2−/− donor T cells. (D) Representative flow cytometric analysis showing upregulation of the activation marker CD25 in the CFSElow population on WT donor T cells. Expression of activation markers at 96 hours (E) and apoptotic marker (F) on WT and Nrf2−/− donor T cells, gated on H2-Kb+CFSElow populations. Data represent the mean + standard deviation combined from 2 or 3 independent experiments (n ≥ 10 per group). *P < .05, **P < .01, ****P < .0001.
Nrf2−/− donor T cells display decreased intestinal infiltration and LPAM-1 expression
Although early activation and proliferation are comparable across all priming sites, different secondary lymphoid organs imprint distinct homing receptor phenotypes and, thus, specify the GVHD target organs to be infiltrated by donor allo-T cells.26,31-36 Antigen-presenting cells in the mesenteric lymph nodes (MLNs) play a role in upregulating gut-homing receptors on donor allo-T cells. We first measured donor T cells on days 7 and 14 after allo-HCT and found significantly more donor Nrf2−/− CD8+ T cells in recipient spleens and equivalent numbers of donor CD4+ and CD8+ T cells in recipient MLNs (Figure 5A-B). However, we observed significantly fewer CD8+ T cells infiltrating the liver and intestinal tract in recipients of Nrf2−/− donor T cells, implying defective trafficking (Figure 5C-D). We further examined donor allo-T cell expression of gut-specific homing molecules, including LPAM-1 (homing to liver and small and large intestines) and CCR9 (small intestine). Although we did not observe any difference in CCR9 expression, we saw a significant decrease in the expression of LPAM-1 on Nrf2−/− donor CD8+ T cells in recipient spleens (Figure 5E-F). Collectively, these data suggest that decreased upregulation of LPAM-1 by donor CD8+ T cells in the recipient spleen could partially explain the decreased infiltration of donor Nrf2−/− allo-T cells in the intestines.
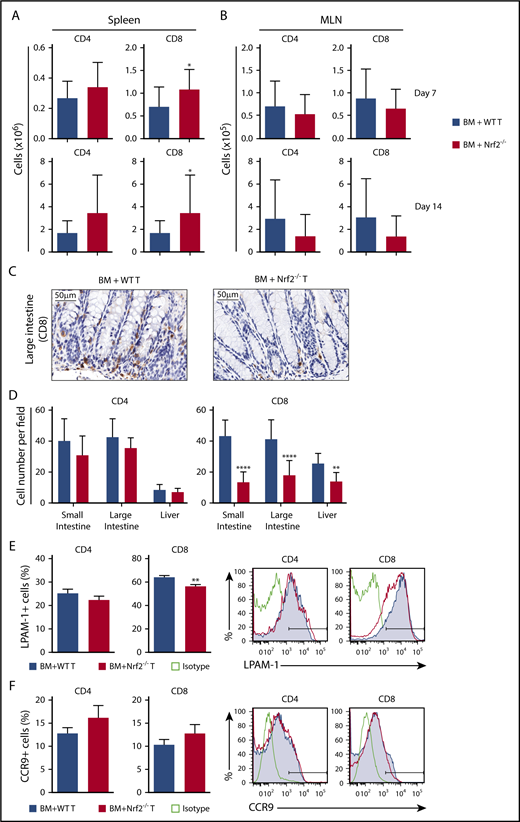
Nrf2−/−donor T cells display decreased intestinal infiltration and LPAM-1 expression. (A-B) Lethally irradiated BALB/c recipients were transplanted with WT B6 TCD BM and 0.5 × 106 B6 WT or Nrf2−/− T cells. Recipient organs were analyzed on days 7 and 14 after HCT. Spleens (n = 15 per group, 3 independent experiments) (A) and MLNs (n = 24 or 25 per group, 5 independent experiments) (B) were analyzed for donor CD4+ and CD8+ T cells using flow cytometry. (C-D) Lethally irradiated BALB/c recipients were transplanted with WT B6 TCD BM and 0.5 × 106 B6 WT or Nrf2−/− T cells. Representative immunohistochemistry images (showing CD8+ cells) (C) and quantification of T cells in recipient liver, small and large intestines (D) on day 14 after HCT (n = 16 per group, 2 independent experiments). Splenocytes were analyzed for the percentage of LPAM-1 (E) and CCR9 (F) expression in donor T cells (n = 20 per group, 4 independent experiments). Data represent the mean + standard deviation combined from independent experiments. *P < .05, **P < .01, ****P < .0001.
Nrf2−/−donor T cells display decreased intestinal infiltration and LPAM-1 expression. (A-B) Lethally irradiated BALB/c recipients were transplanted with WT B6 TCD BM and 0.5 × 106 B6 WT or Nrf2−/− T cells. Recipient organs were analyzed on days 7 and 14 after HCT. Spleens (n = 15 per group, 3 independent experiments) (A) and MLNs (n = 24 or 25 per group, 5 independent experiments) (B) were analyzed for donor CD4+ and CD8+ T cells using flow cytometry. (C-D) Lethally irradiated BALB/c recipients were transplanted with WT B6 TCD BM and 0.5 × 106 B6 WT or Nrf2−/− T cells. Representative immunohistochemistry images (showing CD8+ cells) (C) and quantification of T cells in recipient liver, small and large intestines (D) on day 14 after HCT (n = 16 per group, 2 independent experiments). Splenocytes were analyzed for the percentage of LPAM-1 (E) and CCR9 (F) expression in donor T cells (n = 20 per group, 4 independent experiments). Data represent the mean + standard deviation combined from independent experiments. *P < .05, **P < .01, ****P < .0001.
Abrogation of GVHD in Nrf2−/− allo-T cell recipients is associated with a greater proportion of donor-derived CD4+Helios+ Tregs
We next analyzed whether Nrf2 could affect the polarization of donor CD4+ T cells after allo-HCT. We found a significantly greater percentage and absolute number of donor-derived CD4+ Tregs in the spleens of Nrf2−/− T-cell recipients on day 7 posttransplant (Figure 6A-B). Tregs have been broadly divided into 2 main categories: thymus-derived natural Tregs (nTregs) and peripherally induced Tregs (iTregs). To determine whether Nrf2 regulates the peripheral generation of iTregs, we activated naive CD4+ T cells isolated from steady-state spleens under Treg-differentiating cytokine conditions in vitro and found an equivalent fraction of WT and Nrf2−/− CD4+ T cells polarized toward Tregs in vitro (supplemental Figure 1C-D).
![Figure 6. Abrogation of GVHD in Nrf2−/− allo-T cell recipients is associated with a greater proportion of donor-derived CD4+ Helios+ Tregs. (A-G) Lethally irradiated BALB/c recipients were transplanted with WT B6 TCD BM and 0.5 × 106 B6 WT or Nrf2−/− T cells. Donor CD4+ T-cell subsets in recipient spleens were analyzed on day 7 after HCT. Data represent the mean ± standard deviation. (A) Absolute numbers of WT and Nrf2−/− donor-derived CD4+ Th1 (T-bet+) cells, Th2 (GATA-3+) cells, Th17 (RORγt+) cells, and Tregs (FoxP3+CD25+) (n = 15 per group, combined from 3 independent experiments). (B) Representative flow cytometric dot plots showing the frequency of Tregs within donor CD4+ populations. (C) Representative flow cytometric analysis and bar graphs showing the mean fluorescence intensity (MFI) of intracellular Helios expression within donor-derived Tregs (n = 5 per group, 1 of 2 reproducible experiments). (D) The proportion of donor-derived Tregs expressing Helios (n = 15 per group, combined from 2 independent experiments). (E) Representative flow cytometric analysis and bar graph showing the MFI of intracellular FoxP3 expression within donor-derived Tregs (n = 5 per group, 1 of 3 reproducible experiments). (F) IL-2 levels in the recipient sera were analyzed using Luminex (n = 19 or 20 per group combined from 4 independent experiments). (G) An IL-2 secretion assay was performed to detect IL-2 levels on the donor T-cell surface, and MFI was analyzed using flow cytometry (n = 5 per group, 1 of 2 reproducible experiments). (H) WT CD45.1+ B6 TCD BM plus CD45.2+ WT vs Nrf2−/− T cells were transplanted into lethally irradiated CD45.1+ B6 (syngeneic HCT [syn-HCT]) or BALB/c (Allo-HCT) recipients. Bar graphs show the MFI of CD25 receptor and intracellular Helios expression within donor-derived Tregs. Data represent the mean + standard deviation from 1 of 2 reproducible experiments (n = 5 per group). (I) Lethally irradiated BALB/c recipients transplanted with WT B6 TCD BM alone or with a total of 0.5 × 106 B6 putative Treg (CD4+CD25+)-depleted B6 WT or Nrf2−/− conventional CD4+ and CD8+ T cells (Tcon) were monitored daily for survival. Kaplan-Meier analysis showing combined data from 2 independent experiments. (J) Schematic model of the proposed mechanism by which Nrf2 regulates CD4+ T cells in GVHD. *P < .05, **P < .01, ***P < .005, ****P < .0001.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/132/26/10.1182_blood-2017-10-812941/6/m_blood812941f6.png?Expires=1766645670&Signature=lFG5WaO21KikRya1fdPpLrmt15jlV2V-mzFNDgR0TD0UK7xRVrhgntl-t6skM-IKFQ8aS8KbN3YXB2ccw2GXOjeTpBJdQhfa4Z77tAEK9IOHxdnsRc82O5tlKMTB6Fp4WOhg4UBzzWeNB1-6S~5mGZDHtI8pG3QHOQboHabTH5ipFAwLhA7v8yyYwe-0DyFLNcgdBIp748yv9kYby2oPg3SwgMk9qI1lxHw-STjGpdSIiMKBrjw9BdnmvBdH26m4K3ljX0HmMRTKv35TN9S~SDU3ZOpPCGp4N8L~gmNTq9g9fN2Fi1yoCTGUjPAUN8HUFC5Iy-eDPDa5M972ETN1-g__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Abrogation of GVHD in Nrf2−/−allo-T cell recipients is associated with a greater proportion of donor-derived CD4+Helios+Tregs. (A-G) Lethally irradiated BALB/c recipients were transplanted with WT B6 TCD BM and 0.5 × 106 B6 WT or Nrf2−/− T cells. Donor CD4+ T-cell subsets in recipient spleens were analyzed on day 7 after HCT. Data represent the mean ± standard deviation. (A) Absolute numbers of WT and Nrf2−/− donor-derived CD4+ Th1 (T-bet+) cells, Th2 (GATA-3+) cells, Th17 (RORγt+) cells, and Tregs (FoxP3+CD25+) (n = 15 per group, combined from 3 independent experiments). (B) Representative flow cytometric dot plots showing the frequency of Tregs within donor CD4+ populations. (C) Representative flow cytometric analysis and bar graphs showing the mean fluorescence intensity (MFI) of intracellular Helios expression within donor-derived Tregs (n = 5 per group, 1 of 2 reproducible experiments). (D) The proportion of donor-derived Tregs expressing Helios (n = 15 per group, combined from 2 independent experiments). (E) Representative flow cytometric analysis and bar graph showing the MFI of intracellular FoxP3 expression within donor-derived Tregs (n = 5 per group, 1 of 3 reproducible experiments). (F) IL-2 levels in the recipient sera were analyzed using Luminex (n = 19 or 20 per group combined from 4 independent experiments). (G) An IL-2 secretion assay was performed to detect IL-2 levels on the donor T-cell surface, and MFI was analyzed using flow cytometry (n = 5 per group, 1 of 2 reproducible experiments). (H) WT CD45.1+ B6 TCD BM plus CD45.2+ WT vs Nrf2−/− T cells were transplanted into lethally irradiated CD45.1+ B6 (syngeneic HCT [syn-HCT]) or BALB/c (Allo-HCT) recipients. Bar graphs show the MFI of CD25 receptor and intracellular Helios expression within donor-derived Tregs. Data represent the mean + standard deviation from 1 of 2 reproducible experiments (n = 5 per group). (I) Lethally irradiated BALB/c recipients transplanted with WT B6 TCD BM alone or with a total of 0.5 × 106 B6 putative Treg (CD4+CD25+)-depleted B6 WT or Nrf2−/− conventional CD4+ and CD8+ T cells (Tcon) were monitored daily for survival. Kaplan-Meier analysis showing combined data from 2 independent experiments. (J) Schematic model of the proposed mechanism by which Nrf2 regulates CD4+ T cells in GVHD. *P < .05, **P < .01, ***P < .005, ****P < .0001.
Abrogation of GVHD in Nrf2−/−allo-T cell recipients is associated with a greater proportion of donor-derived CD4+Helios+Tregs. (A-G) Lethally irradiated BALB/c recipients were transplanted with WT B6 TCD BM and 0.5 × 106 B6 WT or Nrf2−/− T cells. Donor CD4+ T-cell subsets in recipient spleens were analyzed on day 7 after HCT. Data represent the mean ± standard deviation. (A) Absolute numbers of WT and Nrf2−/− donor-derived CD4+ Th1 (T-bet+) cells, Th2 (GATA-3+) cells, Th17 (RORγt+) cells, and Tregs (FoxP3+CD25+) (n = 15 per group, combined from 3 independent experiments). (B) Representative flow cytometric dot plots showing the frequency of Tregs within donor CD4+ populations. (C) Representative flow cytometric analysis and bar graphs showing the mean fluorescence intensity (MFI) of intracellular Helios expression within donor-derived Tregs (n = 5 per group, 1 of 2 reproducible experiments). (D) The proportion of donor-derived Tregs expressing Helios (n = 15 per group, combined from 2 independent experiments). (E) Representative flow cytometric analysis and bar graph showing the MFI of intracellular FoxP3 expression within donor-derived Tregs (n = 5 per group, 1 of 3 reproducible experiments). (F) IL-2 levels in the recipient sera were analyzed using Luminex (n = 19 or 20 per group combined from 4 independent experiments). (G) An IL-2 secretion assay was performed to detect IL-2 levels on the donor T-cell surface, and MFI was analyzed using flow cytometry (n = 5 per group, 1 of 2 reproducible experiments). (H) WT CD45.1+ B6 TCD BM plus CD45.2+ WT vs Nrf2−/− T cells were transplanted into lethally irradiated CD45.1+ B6 (syngeneic HCT [syn-HCT]) or BALB/c (Allo-HCT) recipients. Bar graphs show the MFI of CD25 receptor and intracellular Helios expression within donor-derived Tregs. Data represent the mean + standard deviation from 1 of 2 reproducible experiments (n = 5 per group). (I) Lethally irradiated BALB/c recipients transplanted with WT B6 TCD BM alone or with a total of 0.5 × 106 B6 putative Treg (CD4+CD25+)-depleted B6 WT or Nrf2−/− conventional CD4+ and CD8+ T cells (Tcon) were monitored daily for survival. Kaplan-Meier analysis showing combined data from 2 independent experiments. (J) Schematic model of the proposed mechanism by which Nrf2 regulates CD4+ T cells in GVHD. *P < .05, **P < .01, ***P < .005, ****P < .0001.
The Ikaros transcription factor family member Helios and the membrane-bound tyrosine kinase receptor neuropilin-1 are highly enriched in nTregs; thus, their expression can be used to discriminate nTregs from iTregs.37 Although we did not detect any differences in neuropilin-1 expression, we saw a significantly greater percentage of donor-derived Tregs expressing Helios in allo-HCT recipients of Nrf2−/− T cells than in WT T-cell recipients on day 7 after transplant (Figure 6C-D). Moreover, the expression of Helios on a per-cell basis was also significantly higher in donor Nrf2−/− Tregs than in donor WT Tregs after allo-HCT. Notably, we did not observe any baseline differences in the total number or percentage of donor Tregs, or in their Helios expression, in the donor inoculum prior to transplantation (supplemental Figure 1E-F). Importantly, Nrf2−/− donor Tregs displayed equivalent apoptosis and suppressive function upon activation (supplemental Figure 2).
Recent studies have suggested that Helios is required to stabilize the expression of the transcription factor Foxp3 and, by extension, the abundance and inhibitory activity of Tregs in inflammatory settings.38,39 In accordance with this notion, we identified an increase in Foxp3 protein expression within donor Nrf2−/− Tregs in allo-HCT recipients38 (Figure 6E). Recent studies have elucidated that Nrf2 activators suppress CD25 (interleukin-2Ra [IL-2Ra]) expression in human Jurkat T cells and that IL-2 induces selective expansion of Helios+ Tregs from healthy donors ex vivo, as well as in patients with chronic GVHD in vivo.40,41 Thus, we hypothesized that Nrf2 regulates Helios expression through IL-2/CD25 signaling. Interestingly, although we detected comparable levels of IL-2 in the sera or on the T-cell surface of WT and Nrf2−/− allo-T cell recipients, we found a significant increase in CD25 expression in Nrf2−/− donor Tregs on day 7 of allo-HCT (Figure 6F-H). Moreover, the increased expression of Helios and CD25 in Nrf2−/− donor Tregs was observed in allo-HCT settings, but not in syn-HCT settings, suggesting that this regulation depends on recognition of alloantigens (Figure 6H). To further elucidate the biological significance of Nrf2 in nTregs, we removed CD4+CD25+ putative Tregs from the donor inoculum and transplanted only conventional T cells. We found that the survival advantage with donor Nrf2−/− T cells was abolished when Tregs were depleted from the donor inoculum (Figure 6I). Thus, we propose that, upon TCR engagement, Nrf2 in donor CD4+ T cells suppresses CD25 expression and attenuates IL-2 signaling, thereby downregulating Helios and destabilizing Foxp3 expression. Collectively, these effects restrain the expansion of nTregs during GVHD (Figure 6J).
Nrf2−/− CD8+ T cells show intact cytotoxicity and GVT activity
We next analyzed the role of Nrf2 in donor CD8+ T cells independently of its effects on CD4+ T cells. To this end, we activated naive CD8+ T cells isolated from steady-state spleens with TCR ligation in vitro and examined their cytolytic capacity against tumor cells bearing an MHC-disparate alloantigen (A20, murine B cell lymphoma). Nrf2−/− CD8+ T cells displayed intact cytolysis in vitro (Figure 7A). To directly assess their cytotoxicity upon alloantigen activation in an in vivo model, as well as to maintain CD4+ help for CD8+ T-cell alloreactivity, we transplanted WT CD4+ T cells, mixed with WT or Nrf2−/− CD8+ T cells at a physiologic ratio (2:1), into allo-HCT recipients15,42 that were subsequently challenged with fluorescently labeled allogeneic target cells mixed with control syngeneic cells (1:1) on day 7 after allo-HCT.43,44 Compared with the allo-HCT recipients of donor BM cells only (without T cells), recipients of WT and Nrf2−/− CD8+ T cells demonstrated equally efficient elimination of allogeneic target cells in vivo, consistent with our in vitro findings (Figure 7B).
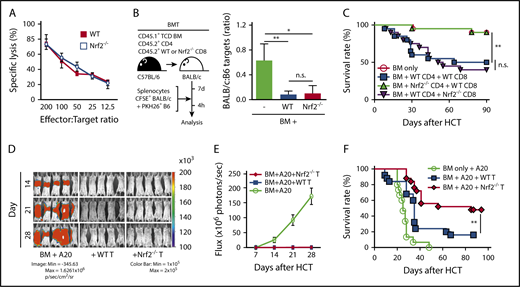
Nrf2 is dispensable for donor CD8+T-cell cytotoxicity and GVT capacity. (A) In vitro cytotoxicity assay. WT and Nrf2−/− CD8+ T cells isolated from steady-state spleens were activated with anti-CD3 and anti-CD28 in vitro for 72 hours, followed by incubation with 51Cr-labeled allogeneic A20 tumor target cells for 18 or 19 hours (n = 2 independent experiments). (B) In vivo cytotoxicity assay. Lethally irradiated BALB/c recipients were transplanted with WT B6 TCD BM and a total of 0.5 × 106 T cells, composed of B6 WT CD4+ cells mixed with WT or Nrf2−/− CD8+ T cells at a 2:1 ratio. On day 7, recipients were challenged with a 1:1 infusion of CFSE-labeled BALB/c allogeneic and PKH26-labeled B6 syngeneic control targets. Elimination of targets was assessed in the spleen by flow cytometry 4 hours after challenge (n = 3 in BM group, n = 9 per group in BM + WT or Nrf2−/− T groups; 2 independent experiments). (C) Lethally irradiated BALB/c recipients were transplanted with WT B6 TCD BM alone or with a total of 0.5 × 106 T cells, composed of B6 WT or Nrf2−/− CD4+ and CD8+ T cells mixed at a 2:1 ratio. Recipients were monitored daily for mortality (n = 20 per group; 2 independent experiments). (D-E) Lethally irradiated BALB/c recipients transplanted with WT B6 TCD BM alone or with 0.5 × 106 WT or Nrf2−/− T cells were challenged with 0.5 × 106 A20-TGL cells. The whole-body distribution of tumor burden was tracked weekly using bioluminescent signal intensity. (D) Representative bioluminescence images showing pseudocolor images superimposed on conventional photographs. (E) Tumor burden in mice quantified as bioluminescence image flux. Data represent mean ± SEM from 2 independent experiments (n = 10 in BM group, n = 18 to 20 each in BM + WT or Nrf2−/− T groups). (F) Lethally irradiated BALB/c recipients transplanted with WT B6 TCD BM alone or with 0.5 × 106 WT or Nrf2−/− T cells were challenged with 0.5 × 106 A20-TGL cells. Recipients were monitored daily for mortality (n = 15 in BM group, n = 25 each in BM + WT or Nrf2−/− T groups; 3 independent experiments). *P < .05, **P < .01. n.s., not significant.
Nrf2 is dispensable for donor CD8+T-cell cytotoxicity and GVT capacity. (A) In vitro cytotoxicity assay. WT and Nrf2−/− CD8+ T cells isolated from steady-state spleens were activated with anti-CD3 and anti-CD28 in vitro for 72 hours, followed by incubation with 51Cr-labeled allogeneic A20 tumor target cells for 18 or 19 hours (n = 2 independent experiments). (B) In vivo cytotoxicity assay. Lethally irradiated BALB/c recipients were transplanted with WT B6 TCD BM and a total of 0.5 × 106 T cells, composed of B6 WT CD4+ cells mixed with WT or Nrf2−/− CD8+ T cells at a 2:1 ratio. On day 7, recipients were challenged with a 1:1 infusion of CFSE-labeled BALB/c allogeneic and PKH26-labeled B6 syngeneic control targets. Elimination of targets was assessed in the spleen by flow cytometry 4 hours after challenge (n = 3 in BM group, n = 9 per group in BM + WT or Nrf2−/− T groups; 2 independent experiments). (C) Lethally irradiated BALB/c recipients were transplanted with WT B6 TCD BM alone or with a total of 0.5 × 106 T cells, composed of B6 WT or Nrf2−/− CD4+ and CD8+ T cells mixed at a 2:1 ratio. Recipients were monitored daily for mortality (n = 20 per group; 2 independent experiments). (D-E) Lethally irradiated BALB/c recipients transplanted with WT B6 TCD BM alone or with 0.5 × 106 WT or Nrf2−/− T cells were challenged with 0.5 × 106 A20-TGL cells. The whole-body distribution of tumor burden was tracked weekly using bioluminescent signal intensity. (D) Representative bioluminescence images showing pseudocolor images superimposed on conventional photographs. (E) Tumor burden in mice quantified as bioluminescence image flux. Data represent mean ± SEM from 2 independent experiments (n = 10 in BM group, n = 18 to 20 each in BM + WT or Nrf2−/− T groups). (F) Lethally irradiated BALB/c recipients transplanted with WT B6 TCD BM alone or with 0.5 × 106 WT or Nrf2−/− T cells were challenged with 0.5 × 106 A20-TGL cells. Recipients were monitored daily for mortality (n = 15 in BM group, n = 25 each in BM + WT or Nrf2−/− T groups; 3 independent experiments). *P < .05, **P < .01. n.s., not significant.
To determine the effect of Nrf2 deficiency in donor CD8+ T cells on GVHD, we transplanted WT CD4+ T cells mixed with WT or Nrf2−/− CD8+ T cells. As expected, we observed similar GVHD mortality in allo-HCT recipients of donor Nrf2−/− and WT CD8+ T cells (Figure 7C) and reduced GVHD mortality in allo-HCT recipients of donor Nrf2−/− cells compared with WT CD4+ T cells. These findings further support the notion that Nrf2 predominantly regulates donor CD4+ T cells and is dispensable for CD8+ T-cell alloactivation and organ damage during GVHD.
Finally, we analyzed the effect of Nrf2 deficiency on the GVT activity of donor T cells in a well-established in vivo GVHD/GVT model: B6 → BALB/c with A20 lymphoma. We found similar GVT activity and A20 clearance in allo-HCT recipients of donor Nrf2−/− and WT T cells (Figure 7D-E). However, tumor-bearing allo-HCT recipients of Nrf2−/− T cells had significantly better overall survival compared with tumor-bearing allo-HCT recipients of donor WT T cells as a result of mitigated GVHD (Figure 7F; Table 1).
Cause of death in GVT experiments
| Group | Tumor, n (%) | GVHD, n (%) | Censored, n (%)* |
|---|---|---|---|
| BM + A20 | 15/15 (100) | 0/15 (0) | 0/15 (0) |
| BM + WT T + A20 | 4/25 (16) | 14/25 (56) | 3/25 (12) |
| BM + Nrf2−/− T + A20 | 2/25 (8) | 9/25 (36) | 2/25 (8) |
| Group | Tumor, n (%) | GVHD, n (%) | Censored, n (%)* |
|---|---|---|---|
| BM + A20 | 15/15 (100) | 0/15 (0) | 0/15 (0) |
| BM + WT T + A20 | 4/25 (16) | 14/25 (56) | 3/25 (12) |
| BM + Nrf2−/− T + A20 | 2/25 (8) | 9/25 (36) | 2/25 (8) |
See Figure 7F for details about the groups. The cause of death was determined by necropsy and histopathology.
Not analyzed because of tissue necrosis.
Discussion
Understanding and dissecting the undesired GVHD and beneficial GVT activities of donor allo-T cells could significantly improve the clinical outcomes of transplant recipients. In this study, we identified a role for Nrf2 in the regulation of donor T cells and their GVHD activity. In the absence of Nrf2 activation, acute GVHD is attenuated by expanded Helios+ nTregs, whereas GVT is preserved by intact CD8+ cytotoxicity.
Nrf2 has been reported to modulate inflammation in different disease models, and Nrf2−/− mice demonstrate increased susceptibility to experimental autoimmune encephalomyelitis and asthma.45,46 However, the ubiquitous nature of Nrf2 means that Nrf2 deficiency in other cells involved in inflammation could have outweighed the regulatory effects of Nrf2 in T-cell activation. Interestingly, a recent study reported that constitutive activation of Nrf2 in T cells protected mice from ischemia-reperfusion–induced acute kidney injury with a higher frequency, but not total number, of intrarenal Tregs, as well as reduced intracellular tumor necrosis factor-α, interferon-γ, and IL-17 in CD4+ T cells. Together with our observations that constitutive inhibition of Nrf2 in T cells ameliorated GVHD, these findings indicate the dual pro- and anti-inflammatory potential of Nrf2 in various experimentally induced inflammatory models. Thus, further exploration and a thorough characterization of the immunomodulatory effects of Nrf2 in a cell type– and model-specific manner is warranted to further its clinical utility.
Earlier studies of the role of Nrf2 in T cells, which focused on the effects of Nrf2 activation on cytokine production by CD4+ subsets in vitro, reported conflicting results.47-49 A major limitation of these studies is the reliance on electrophilic compounds that target multiple pathways. Although the synthetic antioxidant tert-butylhydroquinone, for example, was commonly used to activate Nrf2 in vitro in these studies, it has also been reported to activate the PI3K-Akt pathway and inhibit FoxO3a activity in other cell types.50-52 One group observed an increase in Nrf2 expression when human T cells from healthy donors were activated in vitro.49 However, this process seems to be independent of oxidative stress, because the potent reducing agent N-acetylcysteine, which is known to prevent oxidative stress–dependent activation of Nrf2, failed to inhibit the induction of Nrf2 expression in activated T cells.49 We similarly found increased protein levels and nuclear translocation of Nrf2, yet we did not observe an increase in its canonical antioxidant target genes in activated WT murine T cells in vitro (data not shown). These findings suggest that a noncanonical Nrf2 pathway, upstream and/or downstream, may exist during T-cell activation.
We demonstrated here that alloactivation in vivo enhanced intracellular levels of Nrf2 in donor T cells (especially CD4+). However, this upregulation of Nrf2 during allo-HCT is not required for initial alloactivation, because donor Nrf2−/− T cells displayed intact proliferation, activation, and apoptosis during the early phase of alloreactivity. Instead, Nrf2 exerts its effects during the expansion and target organ–infiltration phase of acute GVHD, during which (day 7 posttransplantation) we noted an increase in donor Helios+ nTregs in allo-HCT recipients of donor Nrf2−/− T cells. Furthermore, we demonstrated that the increase in Tregs resulted from stabilization and/or persistence of nTregs present in the inoculum that were free from regulation by Nrf2 and its proinflammatory properties. This was evidenced by (1) the disappearance of the survival advantage in allo-HCT recipients of donor Nrf2−/− T cells when putative Tregs were removed from the graft before transplant and (2) the relative increase in Helios expression, a marker for nTreg and critical for Foxp3 stability, in donor Nrf2−/− Tregs after, but not before, transplant. This increase in Tregs in the absence of Nrf2 is associated with reduced systemic inflammation after allo-HCT, as shown by the ameliorated GVHD-induced hepatic, intestinal, and thymic damage in Nrf2−/− donor T-cell recipients.
Critical for their potential translation into clinical allo-HCT treatment strategies, Nrf2−/− donor T cells retained their GVT activity. Donor Nrf2−/− CD8+ T cells preserved their cytotoxicity and antitumor activity, and an increased number of CD8+ T cells accumulated in the spleens of allo-HCT recipients of donor Nrf2−/− T cells. One likely contributory mechanism is the decreased upregulation of LPAM-1 in Nrf2−/− CD8+ T cells. This may be secondary to suppression by increased nTregs on antigen-presenting cell priming, which restrains them from infiltrating and damaging GVHD target organs and traps them in the secondary lymphoid organs where lymphohematopoietic tumor cells, such as A20 cells, reside. Nrf2−/− donor CD8+ T cells also possess a higher homeostatic proliferative potential in a lymphopenic environment (data not shown), further contributing to increased numbers of circulating donor Nrf2−/− CD8+ T cells. Moreover, we have previously shown that donor T cells lacking functional LPAM-1 receptors as a result of β7 subunit deficiency have enhanced tumor clearance ability while causing less acute GVHD in MHC-mismatched recipients.16,53 Furthermore, donor Tregs have previously been reported to be dispensable for GVT activity while inhibiting GVHD after allo-HCT.42 Importantly, in addition to GVHD suppression, donor Tregs (as well as CD8+ T cells) have previously been shown to promote hematopoietic stem and progenitor cell engraftment and enhance immune reconstitution.54,55 Collectively, these findings support the development of strategies to suppress the Nrf2 pathway in donor T cells to prevent GVHD while also maintaining GVT in patients undergoing allo-HCT.
Presented in abstract form at the 59th annual meeting of the American Society of Hematology, Atlanta, GA, 9 December 2017.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Jefferson Chan (University of California, Irvine) for providing Nrf2−/− mice. They gratefully acknowledge the technical assistance of Arnab Ghosh, Koichi Takahashi, Hillary V. Jay, Jyotsna Gupta, and Emily Levy, as well as the Animal Imaging Core, Flow Cytometry Core, Molecular Cytology Core, Comparative Pathology Core, and the Research Animal Resource Center at MSKCC.
This work was supported by National Institutes of Health, National Cancer Institute (NCI) grants R01-CA228358-01 (M.R.M.v.d.B.) and R01-CA228308-01 (M.R.M.v.d.B.), Memorial Sloan Kettering Cancer Center Support Grant/Core grant P30 CA008748, and Project 4 of P01-CA023766-39 (M.R.M.v.d.B.) as well as National Institutes of Health, National Heart, Lung, and Blood Institute (NHLBI) grants R01-HL123340-04 (K. Cadwell), K08-HL115355 (A. M. Hanash), and R01-HL125571 (A. M. Hanash). Additional support was received from the National Institutes of Health, National Institute of Aging grant Project 2 of P01-AG052359-02 (M.R.M.v.d.B.) and by the Tri-Institutional Stem Cell Initiative award 2016-013 (S. Rafii). Support was also received from The Lymphoma Foundation, the Susan and Peter Solomon Divisional Genomics Program, the Parker Institute for Cancer Immunotherapy at MSKCC and the European Union's Seventh Framework Programme for Research, Technological Development, and Demonstration under grant agreement 602587. J.J.T. was supported by a Tumor Immunology Predoctoral Fellowship Training Grant from the Cancer Research Institute and the Dorris J. Hutchison Student Fellowship Award from Sloan Kettering Institute. E.V. is a 2018 Amy Strelzer Manasevit Research Scholar. A. M. Hanash also received research support from the Ludwig Center for Cancer Immunotherapy.
Authorship
Contribution: J.J.T. designed and performed the experiments and interpreted the data; E.V., Y.S., K.V.A., and A. M. Holland assisted with experiments, provided substantial intellectual input, and helped to edit the manuscript; O.M.S., L.F.Y., N.L.Y., U.K.R., F.M.K., S.R.L., A.L., S.Y., Y.-Y.F., C. Liu, C. Lezcano, and G.F.M. performed and analyzed experiments and helped to interpret results; I.-K.N., R.R.J., and A. M. Hanash provided substantial intellectual input; J.A.D. and M.R.M.v.d.B. supervised the research; and J.J.T., E.V., and M.R.M.v.d.B. wrote the manuscript.
Conflict-of-interest disclosure: M.R.M.v.d.B. has intellectual property with both Seres Therapeutics (microbiota composition related to GVH disease) and Juno Therapeutics (adoptive cell therapy with precursor T cells), which is not related to the subject matter of this manuscript; has consulted, received honorarium from, or participated in advisory boards for Flagship Ventures, Novartis, Evelo, Seres Therepeutics, Jazz Pharmaceuticals, Therakos, Amgen, Merck & Co. Inc., the Acute Leukemia Forum, and DKMS Medical Council (Board) and research support; and has stock options with Seres Therapeutics. A. M. Hanash has performed consulting for Ziopharm and Nexus Global Group. The remaining authors declare no competing financial interests.
Correspondence: Jennifer J. Tsai, 1275 York Ave, Box 111, New York, NY 10065; e-mail: tsaij1@mskcc.org; and Marcel R. M. van den Brink, 1275 York Ave, Box 111, New York, NY 10065; e-mail: vandenbm@mskcc.org.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal