

Abstract
In 2013, Maxson and colleagues reported mutations in colony stimulating factor-3 (CSF3R) from chronic neutrophilic leukemia (CNL), and atypical CML (aCML) patients. These mutations are clustered into two different regions of the receptor protein; membrane proximal region (proximal mutation) and frameshift or non-sense mutations in the cytoplasmic tail (truncation mutation). Further analysis revealed a significant majority of patients harbor both membrane-proximal and truncation mutations on the same allele (compound mutations). In vivo analysis of these mutants using mouse models revealed that both membrane proximal and compound mutations induce aggressive leukemia while truncation mutations are non-leukemic. Further studies showed that proximal mutation is sensitive to both JAK2 and MAPK inhibitors while compound mutation is only sensitive to MAPK inhibition but resistant to JAK2 inhibition. Unfortunately, unlike imatinib (BCR-ABL inhibitor) both clinically approved JAK2 inhibitor (ruxolitinib) and MEK1/2 inhibitor (trametinib) are not cytotoxic. Instead, they are cytostatic. In other words, both of these inhibitors lack clonal selectivity for the leukemic cells. While treatment with Jak2 or Mek1/2 inhibitors provide some relief to patients, but the responses are short-lived, as leukemic cells in a very short period of time (4-6 months) adapt to thrive under Jak2 and Mek1/2 inhibition resulting to loss of treatment response and disease relapse. In addition, prolonged inhibition of JAK2 and MEK/ERK are seemingly detrimental for normal hematopoietic and adult tissue homeostasis. These observations warrant identifying additional therapeutic targets that must be safe and able to eliminate the mutant clone.
A recent study using CML as a model system to understand the mechanisms driving therapeutic response (clonal selectivity) to TKI (tyrosine kinase inhibitor) treatment demonstrated that the AP1 transcription factor c-Fos and dual specificity phosphatase, Dusp1, regulate the therapeutic response to drug treatments. Perhaps more interestingly, these studies provided evidence that the expression levels of c-Fos and Dusp1 determine apoptotic thresh-hold in cancer cells such that lower levels confer sensitivity, while higher levels drive resistance to TKI treatments (Kesarwani M, et al. Nature Medicine, 2017).
Results
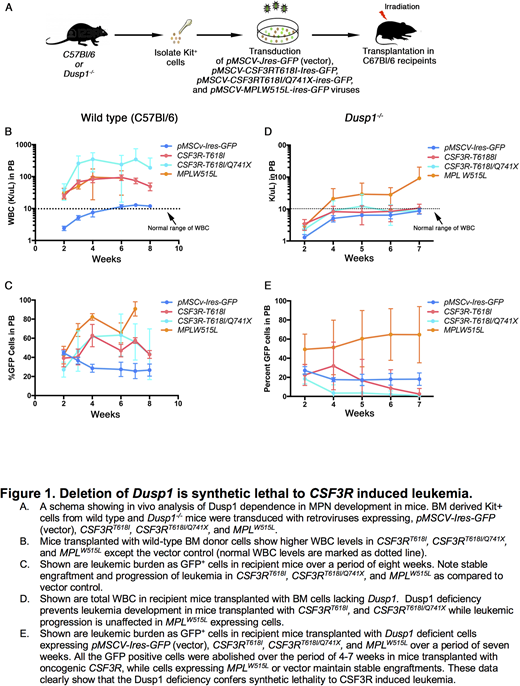
We hypothesized that CSF3R induced CNL might have higher expression of c-Fos and Dusp1 resulting to a higher apoptotic threshold, and thus, abrogating the cytotoxic effect of Jak2 and Mek1/2 inhibitors. To test this, we performed gene expression analysis of bonemarrow cells expressing proximal, truncation, compound mutations and pMSCV-Ires-GFP (as a control). These analyses revealed elevated 4-8-fold overexpression of Dusp1 in cells expressing membrane proximal and compound CSF3R mutations in comparison to vector control and non-leukemic truncation mutations. Further genetic studies using Dusp1 knock-out mice showed dependence of oncogenic CSF3R (proximal and compound) on the Dusp1. Genetic deletion of Dusp1 in cells expressing oncogenic CSF3R (proximal and compound) is synthetic lethal (Fig. 1). Mice transplanted with CSF3RT618I (proximal mutation) or CSF3RT618I/Q741X (compound mutation) expressing cells lacking Dusp1 show gradual depletion of leukemic cells where leukemic cells are completely cleared within 4-7 weeks (Fig. 1E). Rate of clearance is much faster in compound mutation, cells are cleared within 4 weeks after the transplantation (Fig. 1E). In contrast, mice transplanted with cells expressing pMSCV-Ires-GFP (vector) and MPLW515L (another receptor-oncogene induces myeloproliferative neoplasm) show stable engraftment. These data provide evidence that the oncogenic CSF3R is uniquely addicted to Dusp1 and crucial for disease development. Therefore, Dusp1 targeting in CSF3R mutated leukemias may exert a stable and curative response.
Conclusion.
We observed Dusp1 overexpression in CNL expressing oncogenic CSF3R (proximal and compound), but not in cells expressing non-oncogenic CSF3R (truncation mutations). Oncogenic CSF3R is uniquely addicted to the DUSP1 expression, where genetic deletion of DUSP1 is lethal to CSF3R driven leukemias but not to the other leukemic drivers, such as, MPLW515L. Thus, providing a rationale for targeting the DUSP1 in CSF3R driven CNL to eliminate the mutant clones for curative response.
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal