

Abstract
Background: Hurler's syndrome is the most severe phenotype of Mucopolysaccharidosis 1 (MPS1H) with severe deficiency of the lysosomal enzyme alpha-L-iduronidase causing heparan and dermatan sulfate accumulation, CNS and somatic manifestations and premature death. Haematopoeitic Stem Cell Transplant (HSCT) alters the disease phenotype and prevents early death, and is standard of care in this condition. The use of an established 'donor hierarchy' recommending Unrelated Cord Blood Transplantation (UCBT) in the absence of a HLA-matched, non-carrier sibling, has resulted in improved engrafted and surviving rates and better post-enzyme levels with improved somatic and CNS clinical outcomes. However, UCBT for malignant and non-malignant conditions has led to high rates of post-transplant autoimmune disease, most frequently Autoimmune Cytopenia (AIC). We report a large cohort of MPS1H patients receiving busulfan-based UCBT and show that AIC is a frequently encountered complication of treatment with significant associated morbidity and we identify important risk factors for its occurrence.
Methods: This was a retrospective observational study of patients undergoing first UCBT for Hurler's Syndrome at the Royal Manchester Children's Hospital between 30th September 2004 and 15th March 2018. An episode of AIC was defined as an episode of single or multi-lineage cytopenia occurring post-engraftment, confirmed by the presence of antibodies or diagnosed on the basis of clinical, biochemical and pathologic features. All patients received pK targeted busulfan to achieve myeloablative levels between 80-90 mg/L x hr. Conditioning between 2004 and May 2010 was with Busulfan and Cyclophosphamide (BuCy), and Fludarabine and Busulfan (FluBu) thereafter based on the reported equivocal efficacy and favourable toxicity profile of FluBu conditioning. All patients received serotherapy with Anti-Thymocyte Globulin (ATG) 10mg/kg for In Vivo T-Cell depletion. Timing of ATG was either distal (day -4 to -1) or proximal (day - 9 to -6) in accordance with international consensus at the time. Variables assessed in statistical analysis were conditioning drugs, ATG timing, Absolute Lymphocyte Count pre-conditioning, age-at-transplant, Graft-versus-Host Disease (GVHD) and Total Nucleated Cell dose.
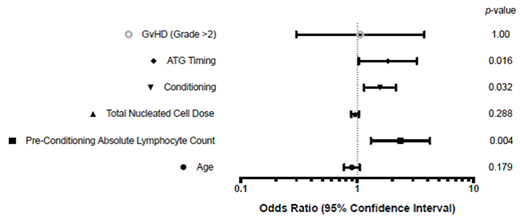
Results and Discussion: Thirty-six patients underwent first UCBT for Hurler's Syndrome. There were 8 episodes of AIC (22%), with median onset post-UCBT of 66 days (range 22-236 days). The median number of therapies required was 4 (range 0-8), with duration of illness ranging from 10 to 215 days. There were no deaths, though there were 2 episodes of life-threatening bleeding. Absolute Lymphocyte Count pre-conditioning (p=0.004), FluBu conditioning (p=0.03) and use of proximal-ATG (p=0.016) were significant risk factors for AIC in univariate analysis(see figure 1). Absolute Lymphocyte Count pre-conditioning was the most significant predictor in multivariable analysis (AOR 5.796, CI 0.921-36.474). This confirms that AIC is a frequent, serious but survivable complication of UCBT for Hurler's Syndrome. This data indicates that the balance between recipient immunity and graft immunity impacts AIC risk and severity, mandating a larger international review of registry data to define the frequency of AIC in this population, and an analysis of the risk factors that predispose to it. By better defining these risk factors, we can identify patients in whom conditioning might be altered to reduce risk.
Figure 1. Univariate analysis of variables predicting an episode of Autoimmune Cytopenia. ALC (p=0.004), ATG timing (0.016) and Conditioning drugs (p=0.032) were significant risk factors. GVHD= Graft-versus-Host Disease ATG= Anti-Thymocyte Globulin
Wynn:Orchard SAB: Membership on an entity's Board of Directors or advisory committees; Orchard Therapeutics: Equity Ownership; Chimerix: Research Funding; Genzyme: Honoraria; Bluebird Bio: Consultancy; Orchard Therapeutics: Consultancy; Chimerix: Consultancy.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal