

Abstract
Background: Acute myeloid leukemia (AML) with complex karyotype (CK) is associated with adverse prognosis with current therapies, in particular in the presence of TP53 mutations. Identification of novel therapeutic strategies is therefore urgently needed for this subgroup of patients.
Methods and aims: As part of the Leucegene project, we RNA sequenced a cohort of 415 clinically annotated primary human AML specimens. Comparative transcriptomic analysis of the 68 CK AML specimens with the non-CK samples of the cohort was performed to identify features associated with the CK AML subgroup. Based on these results, we performed a chemical screen aimed at identifying active agents toward CK AML. Gene expression analysis, primary AML specimen culture and chemical screening were performed as previously described by our group (Lavallée et al., Nature Genetics, 2015; Pabst et al., Nature Methods, 2014; Simon et al., Clinical cancer research, 2017).
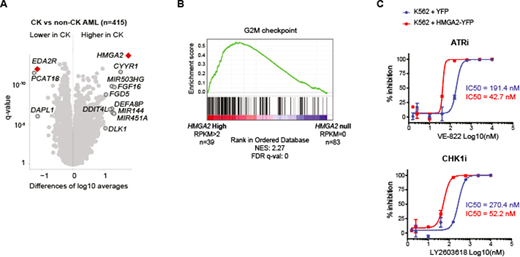
Results: CK AML is a heterogeneous disease by definition and accordingly, Multidimensional Scaling (MDS) analysis fails to identify a distinct transcriptional signature. Nonetheless, comparative transcriptome analysis of CK and non-CK AML specimens revealed highly deregulated genes between these subgroups (Figure 1A). Among them, EDA2R, a known target to p53, is specifically downregulated in CK AML. This reflects the frequent TP53 alterations observed in CK AML. Most importantly, we found that the recently identified AML prognostic gene HMGA2 is overexpressed in the majority of these patients, independently of the TP53 status. HMGA2 is an oncofetal gene expressed in hematopoietic stem cell (HSC) -enriched populations while its expression decreases in progenitors and is nearly absent in mature cells. Interestingly, Gene Set Enrichment Analysis (GSEA) revealed an enrichment for HSC genes in AML specimens with high HMGA2 expression levels, which could reflect a stem-cell origin or a more immature state for these leukemias. Global transcriptomic analysis of CK AML also identified a G2/M checkpoint signature for HMGA2-high specimens (Figure 1B). Accordingly, we observed that chemical inhibitors of the G2/M regulators ATR, CHK1 and WEE1 are preferentially active on HMGA2-high CK specimens and that engineered overexpression of HMGA2 in different leukemia cell lines enhanced sensitivity to G2/M inhibition (Figure 1C). This specific inhibition was independent of the TP53 status.
Conclusions: Here we report a comprehensive transcriptomic analysis of the CK AML specimens of the Leucegene cohort and identified aberrant expression of the HMGA2 oncogene in close to 80% of these samples. Chemical interrogation of primary AML specimens revealed a TP53-independent but HMGA2-mediated sensitization of CK AML to G2/M checkpoint inhibition. Thus, our results reveal a novel vulnerability for AML expressing high levels of HMGA2 and identify potential biological targets for these high-risk patients. Since several CHK1 and PLK1 inhibitors have been or are currently under evaluation in clinical trials, our findings suggest that HMGA2-high AML patients could benefit from this therapeutic approach.
Figure 1: A/ Volcano plot representing the genes differentially expressed in CK versus non-CK AML. Samples with low group median expression (<0.1 RPKM) were removed from this representation. B/ Highest enrichment found by Gene Set Enrichment Analysis comparing HMGA2 high (RPKM>2; n=39) and HMGA2 null (RPKM=0; n=83) AML specimens. C/ Dose-response curves and IC50 values for representative compounds in K562 cells infected with control YFP vector (blue) or HMGA2-YFP expressing vector (red).
Sauvageau:ExCellThera: Employment, Equity Ownership.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal