

In this issue of Blood, 1 apply whole-genome and transcriptome analysis, with sophisticated computational biology to compare the molecular landscape of pediatric Burkitt lymphoma (BL).
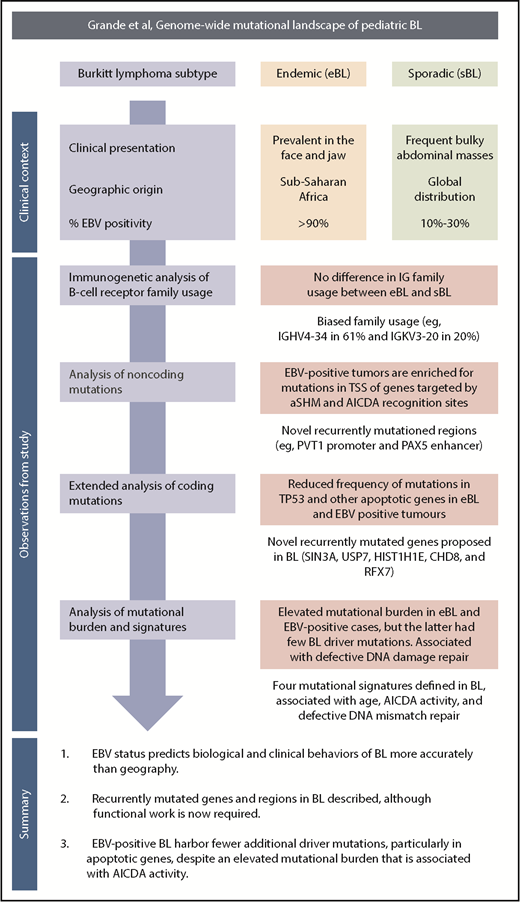
A schematic overview of the clinical context, study observations, and main findings. TSS, transcription start site.
A schematic overview of the clinical context, study observations, and main findings. TSS, transcription start site.
BL is a clinically aggressive, highly proliferative tumor arising from germinal center B cells.2 Characteristic features include a distinctive morphological and histological appearance with immunoglobulin (IG) gene translocations, resulting in constitutive expression of C-MYC through juxtapositioning with the immunoglobulin enhancer.3 Recent genomic and transcriptomic studies have implicated a number of genomic mutations affecting pathways that promote cell growth and proliferation and counteract the apoptotic drive associated with high levels of C-MYC expression.4 The clinical and biological heterogeneity within BL is reflected in the World Health Organization classification, which recognizes 3 variants based on clinical presentation, geographical distribution, and associations with infection and immune status. Endemic BL (eBL) is the commonest childhood malignancy in equatorial regions in which falciparum malaria is holoendemic. Sporadic BL (sBL) occurs worldwide and comprises 1% to 2% of all lymphomas and 30% to 50% of childhood lymphomas in the United States and Western Europe. Immunodeficiency-associated BL (ID-BL) most frequently occurs early in the course of HIV infection. Epstein-Barr virus (EBV) is present in the genome of all tumor cells in >95% of eBL, 20% to 30% of sBL, and 25% to 40% of ID-BL.5 EBV gene expression in BL is typically limited to EBNA1, microRNAs of the BART family, and noncoding EBER RNAs, although LMP2 expression has been documented in some cases. Several studies have noted differences in the transcriptome and/or genomic landscape of BL based on clinical subtype, age, and EBV status, but the precise roles of EBV infection in the pathogenesis of BL are still to be elucidated.6
The authors draw on a large and clinically well-annotated cohort of both eBL and sBL tumor subtypes (see figure).1 The inclusion of “discordant” EBV-negative eBL and EBV-positive sBL cases enabled the authors to test their principal hypothesis; namely, that high-throughput sequencing approaches might provide additional insights into the relationship between genomic abnormalities, EBV status, and clinical variant status. As in normal germinal center B cells, BL cells express activation-induced cytidine deaminase (AICDA), which mediates somatic hypermutation (SHM) of immunoglobulin variable genes and class switch recombination. AICDA can cause off-target mutations in genes outside the immunoglobulin loci (aberrant [a] SHM), and in BL, aberrant class switch recombination is responsible for the IG-MYC translocations. The authors provide compelling evidence linking EBV status with AICDA expression and both the number and the nature of genomic mutations. First, they investigated the underlying mutation processes, inferred from the mutational signatures present in the whole-genome sequencing data. This analysis identified 4 signatures, one of which was associated with AICDA and polymerase η activity (COSMIC signature 9). This signature was strictly associated with AICDA expression and with EBV-positive tumors or eBL. Second, they found that although the total mutational load per genome was greater in eBL or EBV-positive tumors, the prevalence was lower for recurrent coding mutations in genes implicated in BL biology. This difference was most marked for TP53 mutations and for mutations in any apoptotic genes. Importantly, tumor EBV status significantly outperformed clinical variant status in predicting the mutation status of the apoptosis pathway in discordant cases, supporting the notion that EBV abrogates normal apoptotic control in BL. Finally, the authors identified an enrichment of mutations in the transcription start sites of genes known to be affected by aSHM, and in the recognition sites for AICDA. Analysis of gene transcription demonstrated that genes targeted by aSHM were more highly expressed in EBV-positive BL, and that AICDA gene expression correlated with the number genes harboring aSHM, and with the EBV status of the tumor. Of those recurrently mutated genomic regions that were not a target of aSHM, several showed tangible links to genes of known importance in B-cell tumor biology, such as mutations in the PVT1 promoter, a known target of MYC,7 and in the distal enhancer of the B-cell transcription factor, PAX5, similar to those identified in chronic lymphocytic leukemia.8 As AICDA recognition sites were mutated in the PAX5 enhancer and the PVT1 promoter, this raised the tantalizing possibility that AICDA is contributing to eBL pathogenesis by the introduction of noncoding mutations in key regulatory elements.
Interestingly, an additional mutational signature (COSMIC signature 9) associated with defective DNA mismatch repair was more common in EBV-positive cases and eBL but not associated with AICDA expression, possibly implicating a second mechanism whereby EBV may influence the BL genome. Other findings not linked to EBV status included the identification of previously unreported recurring mutations and confirmation of the biased IGHV gene usage previously reported,9 with IGHV4-34 and IGKV3-20 the most frequently used genes.
A key question is the potential clinical relevance of these findings. BL is curable in children and younger adults using multiagent intensive chemotherapy in association with central nervous system prophylaxis and optimal supportive care regardless of the transcriptome and genomic landscape. These regimens are too toxic for older patients, who fare better with lower-dose continuous infusion regimens. However, neither approach is suitable for the management of eBL in resource-poor regions. Less toxic, affordable treatments are urgently required for eBL, older patients with sBL, and the minority of younger sBL patients with relapsed or refractory disease and would be welcomed by younger patients faced with the prospect of intensive chemotherapy. The authors highlight a number of possible therapeutic targets, including BCR signaling inhibitors, a CDK4/6 inhibitor for cases with CCND3 mutations, and EBV.
Additional larger genomic and transcriptomic studies, which include adult patients and are supported by functional analyses, will be necessary, but a recent more empirical, pharmacological profiling approach using BL cell lines indicated synergy between a CDK2/7/9 and bromodomain and extraterminal domain inhibitor, providing encouragement that novel agents may have clinical utility.10 However, the relevance of these approaches to children with eBL is questionable, given the geographic environment where these tumors predominate and the level of health care resource needed to support the prolonged use of these novel agents.
Conflict-of-interest disclosure: The authors declare no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal