

Abstract
We now have the potential to undertake detailed analysis of the inner workings of thousands of cancer cells, one cell at a time, through the emergence of a range of techniques that probe the genome, transcriptome, and proteome combined with the development of bioinformatics pipelines that enable their interpretation. This provides an unprecedented opportunity to better understand the heterogeneity of chronic lymphocytic leukemia and how mutations, activation states, and protein expression at the single-cell level have an impact on disease course, response to treatment, and outcomes. Herein, we review the emerging application of these new techniques to chronic lymphocytic leukemia and examine the insights already attained through this transformative technology.
Introduction
A defining feature of chronic lymphocytic leukemia (CLL) is its vast clinical variability. Although we have gained important understanding of the differences in disease behavior through the use of prognostic schema based on the Rai and Binet classification systems,1,2 immunoglobulin heavy chain variable (IGHV) region mutational status,3-5 and cytogenetic analyses,6 we still have incomplete knowledge of the underlying biologic features that contribute to the tempo of disease, sensitivity, and response to therapy, and to disease progression and relapse.
CLL has consistently been at the forefront of genomic characterization, with massively parallel sequencing studies of CLL first reported in 2011.7-9 Since then, a growing series of studies using sequencing-based technologies have provided us with a new appreciation of the underlying genetic complexity of this disease. To date, hundreds of CLL samples have been subjected to genomic sequencing and analysis, collectively trailblazing the path to discovery of novel CLL driver mutations, detection of clonal evolution, and defined transcriptional and epigenomic signatures.7-14 CLL has been particularly well-suited to this approach because large numbers of pure malignant cells can be readily procured via venipuncture. Moreover, because of the typically indolent course of this disease (characterized by long periods of observation punctuated with treatment), longitudinal sample collection from individual patients is feasible. Until now, the vast majority of these landmark genomic discoveries have been based on the analysis of bulk leukemia but, by definition, this approach averages data from an entire population of potentially heterogeneous individual cells. Hence, important aspects of disease biology can be lost. Single-cell approaches for the study of the genome, transcriptome, or proteome therefore provide an opportunity to study malignant disease at a resolution not possible with bulk analysis.
For CLL, an appreciation of the clinical and biologic insights that can be gained from single-cell analysis has been longstanding. Indeed, single-cell approaches such as karyotyping and fluorescence in situ hybridization (FISH) have been established since the 1960s and remain in routine clinical use.6 Flow cytometry, another single-cell approach and a workhorse of modern clinical laboratories, is routinely used for diagnosis and provides useful prognostic information through assessment of CD38,3 CD49d,15 and ZAP70.16 It also enables response monitoring, including detection of minimal residual disease at high sensitivity, which is important because of its association with inferior outcomes.17-19
The development of higher-dimensional single-cell techniques, especially sequencing-based approaches, now provides the ability to broadly interrogate a larger number of variables, creating new and unprecedented opportunities for in-depth study of the unique aspects of individual cell biology. Although these technologies are transformative (extensively reviewed elsewhere20-28 ), until recently, they were technically laborious, which limited analyses to tens or up to a few hundred single cells. However, recent advances in molecular biology, microfluidics, and droplet-based technologies combined with a rapidly expanding arsenal of techniques with automated workflows and data analysis have now created the opportunity to feasibly characterize tens of thousands of cells from individual samples. For CLL, application of this new emerging technology promises to provide the next major step forward in our understanding of this biologically heterogeneous disease.
Application and considerations of single-cell technology in CLL
A prerequisite for single-cell analysis is the generation of a single-cell suspension, which is relatively straightforward for CLL because suspension cells can readily be accessed from blood and marrow by venipuncture and marrow biopsy. CLL also commonly exists in the lymph node, which can be sampled by biopsy and from which single cells can be obtained through standard tissue disaggregation techniques (Figure 1). To date, most studies of CLL have focused on circulating leukemic cells because of the ease of access of this tissue from blood. However, bulk analysis has revealed differences in CLL gene expression between blood and lymphoid tissues, especially related to B-cell receptor signaling and phosphorylation of downstream targets,29,30 which can be further dissected at the single-cell level. Of potentially equal interest is the parallel assessment of the supporting nonleukemic cells within these tissue compartments, given the potent prosurvival signals they provide to CLL.31-34 Thus, further advances in understanding CLL disease biology can be gained by assessing both the closely apposed leukemic and nontumor cells from within any of the 3 hemato-lymphoid compartments (Figure 1).

Application of single-cell technology to CLL. CLL is present within blood, lymph nodes, and bone marrow where it coexists with a range of immune and stromal cells that are central to disease pathogenesis. Analysis of CLL may focus uniquely on leukemia cells, supporting cells, or both from all 3 distinct tissue compartments. A range of established and developing single-cell techniques characterizing the genome, transcriptome, and proteome have been developed to allow enhanced appreciation of CLL biology. Ab, antibody; lncRNA, long non-coding RNA; miRNA, microRNA; NK, natural killer; scRRBS, single-cell reduced representation bisulfite sequencing; seq, sequencing; WES, whole-exome sequencing; WGS, whole-genome sequencing.
Application of single-cell technology to CLL. CLL is present within blood, lymph nodes, and bone marrow where it coexists with a range of immune and stromal cells that are central to disease pathogenesis. Analysis of CLL may focus uniquely on leukemia cells, supporting cells, or both from all 3 distinct tissue compartments. A range of established and developing single-cell techniques characterizing the genome, transcriptome, and proteome have been developed to allow enhanced appreciation of CLL biology. Ab, antibody; lncRNA, long non-coding RNA; miRNA, microRNA; NK, natural killer; scRRBS, single-cell reduced representation bisulfite sequencing; seq, sequencing; WES, whole-exome sequencing; WGS, whole-genome sequencing.
After a single-cell suspension is generated, individual cells can be analyzed with established techniques, such as karyotypic analysis, FISH, and flow cytometry, or a variety of emerging technologies that probe the genome and epigenome (including methylation and chromatin assembly). Other transcriptome-wide assessment techniques include evaluation of miRNA35 and lncRNA36,37 species that have important roles in CLL biology. Recent work has demonstrated the ability to assess the single-cell proteome at higher resolution through mass cytometry38 or oligonucleotide-tagged antibodies39 that can detect 40 to 80 antigens per cell, respectively (Figure 1).
The currently available single-cell techniques have a spectrum of characteristics that makes some of them better suited for discovery and others better suited for validation (Figure 2). Discovery-based approaches, such as whole-genome sequencing (WGS), whole-exome sequencing (WES), and whole transcriptome sequencing (RNA-seq), capture information on a global level. Although this has the potential to be unbiased by aiming to sequence all of the DNA or messenger RNA (mRNA) contained within a cell, the minute amount of starting material (a few picograms of DNA or RNA) necessitates amplification steps that can result in bias, low coverage, and introduction of errors.20 Of particular importance is the concept of dropouts in which DNA sequences and/or mRNA transcripts are not reliably detected, which cannot be overcome by increasing the sequencing depth because this provides limited additional information.40 Single-cell data also suffer from higher levels of technical and biological noise, with lower and more variable detection compared with bulk approaches because of the inherent differences in individual cells, especially at the transcript level. Furthermore, to detect significant differences between individual cells, larger numbers of cells need to be analyzed. Although this is now possible by using droplet-based techniques, care has to be taken to minimize doublets and triplets, which have the potential to skew results.
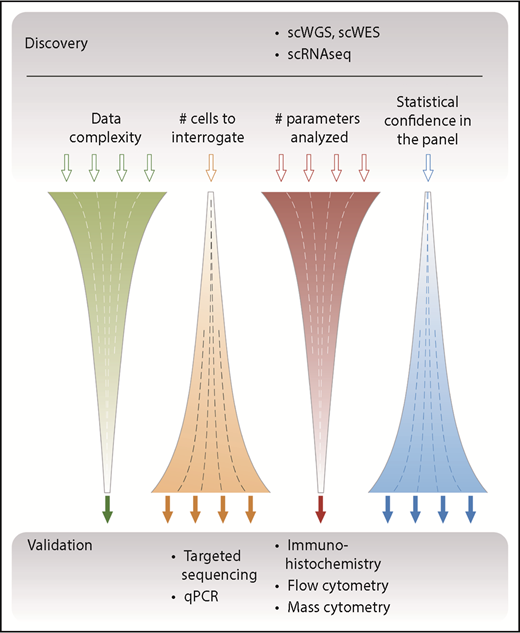
Overview of single-cell–based discovery vs validation approaches. Single-cell approaches such as WGS or WES and single-cell RNA sequencing (scRNAseq) aim to capture information across several parameters and to yield an unbiased global assessment of the genome and transcriptome; hence, they are suitable for discovery. Because of the relatively high cost of generating these complex data, a typical study design focuses these assays on relatively few samples composed of a limited number of single cells. Conversely, targeted approaches use defined primers or antibodies to screen a limited number of parameters but with high sensitivity and statistical confidence. These assays have typically been used as a validation approach, which has the advantage of assessing many more independent patient samples. qPCR, quantitative polymerase chain reaction; scWGS, single-cell WGS.
Overview of single-cell–based discovery vs validation approaches. Single-cell approaches such as WGS or WES and single-cell RNA sequencing (scRNAseq) aim to capture information across several parameters and to yield an unbiased global assessment of the genome and transcriptome; hence, they are suitable for discovery. Because of the relatively high cost of generating these complex data, a typical study design focuses these assays on relatively few samples composed of a limited number of single cells. Conversely, targeted approaches use defined primers or antibodies to screen a limited number of parameters but with high sensitivity and statistical confidence. These assays have typically been used as a validation approach, which has the advantage of assessing many more independent patient samples. qPCR, quantitative polymerase chain reaction; scWGS, single-cell WGS.
In general, the large amount of data generated per cell multiplied by the total number of cells analyzed yields highly complex data, which require concerted bioinformatic analysis to fully resolve.41,42 By contrast, targeted approaches interrogate predetermined loci of interest and thus focus on fewer variables, which enables higher sensitivity and facilitates the analysis of larger numbers of patient samples (and thereby provides greater statistical confidence). An appreciation of these differences and consequent advantages and disadvantages is important not only when selecting the single-cell technique to be used but also when analyzing the subsequent data output.
Application of single-cell analyses to CLL
Increasing numbers of studies have begun to explore the genomic, epigenetic, and transcriptional landscape in CLL at a single-cell level, as summarized in Table 1. Herein we highlight the insights they have provided.
Summary of single-cell studies in CLL
| Reference | Sample input | Methodology | Insight |
|---|---|---|---|
| Zhao et al49 | 116 CLL cells from 2 time points | Whole-genome DNA amplification in individual PCR tubes | sCNA identified and subclonal architecture determined |
| Wang et al50 | 1152 CLL cells from 5 patients | Plate-based targeted multiplex PCR assay following whole-genome amplification | sCNA and sSNV identified and subclonal architecture determined |
| Landau et al58 | 393 CD19+ B cells from 2 healthy donors and 111 CLL cells from 1 patient | Multiplexed scRRBS | Uniformly high proportion of discordant reads in CLL cells compared with normal B cells |
| Chaligne et al59 | 383 CD19+ B cells from 4 healthy donors and 324 CLL cells from 3 patients | Multiplexed scRRBS | Epigenetic phylogenetic tree reconstruction |
| Chaligne et al59 | 42 CLL cells | Multiplexed scRRBS and scRNA sequencing | Increasing phylogenetic distance correlates with decreasing transcriptional similarity |
| Zhao et al49 | 362 CLL cells from 5 time points (35-126 cells per time point) | Plate-based Smart-Seq2 scRNA sequencing | Identification of 6 transcriptional clusters and their evolution over time and with treatment |
| Wang et al50 | 289 CLL cells from 4 patients | Fluidigm C1 System Smart-Seq scRNA sequencing | Using pathway and gene set overdispersion analysis identified unique biological processes demonstrating transcriptional heterogeneity |
| Wang et al44 | 845 CLL cells from 6 patients | Plate-based targeted qPCR using Fluidigm BioMark | Single-cell correlation of alternative splicing variants with SF3B1 mutational status |
| Landau et al13 | 310 CLL cells from 4 patients | Fluidigm C1 System Smart-Seq scRNA sequencing | Correlational of single-cell transcriptional heterogeneity with methylation data |
| Burger et al12 | 473 cells from 3 time points | Plate-based targeted qPCR using Fluidigm BioMark | Mutation detection to determine subclonal architecture |
| Burger et al12 | Polydimethylsiloxane microfluidic device and targeted qPCR | High-sensitivity mutation detection, including demonstration of the presence of rare resistant cells before ibrutinib initiation |
| Reference | Sample input | Methodology | Insight |
|---|---|---|---|
| Zhao et al49 | 116 CLL cells from 2 time points | Whole-genome DNA amplification in individual PCR tubes | sCNA identified and subclonal architecture determined |
| Wang et al50 | 1152 CLL cells from 5 patients | Plate-based targeted multiplex PCR assay following whole-genome amplification | sCNA and sSNV identified and subclonal architecture determined |
| Landau et al58 | 393 CD19+ B cells from 2 healthy donors and 111 CLL cells from 1 patient | Multiplexed scRRBS | Uniformly high proportion of discordant reads in CLL cells compared with normal B cells |
| Chaligne et al59 | 383 CD19+ B cells from 4 healthy donors and 324 CLL cells from 3 patients | Multiplexed scRRBS | Epigenetic phylogenetic tree reconstruction |
| Chaligne et al59 | 42 CLL cells | Multiplexed scRRBS and scRNA sequencing | Increasing phylogenetic distance correlates with decreasing transcriptional similarity |
| Zhao et al49 | 362 CLL cells from 5 time points (35-126 cells per time point) | Plate-based Smart-Seq2 scRNA sequencing | Identification of 6 transcriptional clusters and their evolution over time and with treatment |
| Wang et al50 | 289 CLL cells from 4 patients | Fluidigm C1 System Smart-Seq scRNA sequencing | Using pathway and gene set overdispersion analysis identified unique biological processes demonstrating transcriptional heterogeneity |
| Wang et al44 | 845 CLL cells from 6 patients | Plate-based targeted qPCR using Fluidigm BioMark | Single-cell correlation of alternative splicing variants with SF3B1 mutational status |
| Landau et al13 | 310 CLL cells from 4 patients | Fluidigm C1 System Smart-Seq scRNA sequencing | Correlational of single-cell transcriptional heterogeneity with methylation data |
| Burger et al12 | 473 cells from 3 time points | Plate-based targeted qPCR using Fluidigm BioMark | Mutation detection to determine subclonal architecture |
| Burger et al12 | Polydimethylsiloxane microfluidic device and targeted qPCR | High-sensitivity mutation detection, including demonstration of the presence of rare resistant cells before ibrutinib initiation |
PCR, polymerase chain reaction; qPCR, quantitative PCR; sCNA, somatic copy number alteration; scRNA, single-cell RNA; scRRBS, single-cell reduced representation bisulfite sequencing; sSNV, somatic single nucleotide variant.
DNA-based interrogation
Recent large-scale bulk genomic sequencing of CLL cells has been revolutionary in the identification of driver mutations such as within SF3B1 and NOTCH1,7-9,43 while consequently allowing investigation of their downstream functional significance.44-46 Collectively, these studies have generated a compendium of CLL-associated somatic copy number alterations (sCNAs) and somatic single nucleotide variants (sSNVs). More recently analysis of circulating tumor DNA provides the opportunity to identify mutations in both circulating CLL cells and the tumor microenvironment.47 With these approaches, although it is possible to computationally estimate the cancer cell fraction of these alterations10,48 (ie, the proportion of cells within a population carrying that mutation), these bulk analyses cannot directly distinguish the exact clonal architecture of the cell population, nor can they dissect whether mutations either track together or represent unique parallel progeny events (Figure 3A).
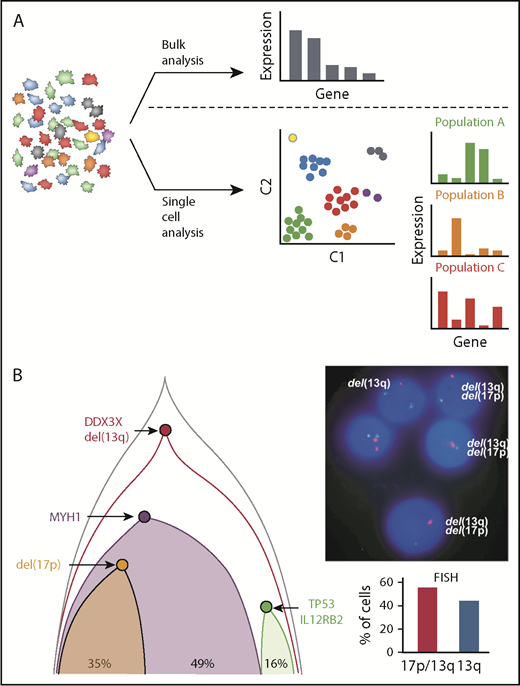
Novel insights into CLL based on single-cell analysis. (A) Bulk analysis of heterogeneous tumor samples, including those admixed with immune and stromal cells, provides useful global gene expression data but results in loss of granularity. Single-cell approaches allow analysis within individual cells as well as in similar cell populations that have clustered to identify unique signatures, cellular processes, networks, and rare cell populations, which provides higher resolution biological insights. (B) Single-cell mutational analysis can yield detailed phylogenetic trees to identify which mutations occur in specific unique subclones and which track together. In this example, multiple sCNA and sSNV were interrogated by using targeted sequencing to establish the underlying clonal architecture of the leukemia. This demonstrated convergent loss of TP53 with 17p deletion in 1 subclone and TP53 mutation in another, which was further corroborated by conventional FISH analysis by Wang et al.50
Novel insights into CLL based on single-cell analysis. (A) Bulk analysis of heterogeneous tumor samples, including those admixed with immune and stromal cells, provides useful global gene expression data but results in loss of granularity. Single-cell approaches allow analysis within individual cells as well as in similar cell populations that have clustered to identify unique signatures, cellular processes, networks, and rare cell populations, which provides higher resolution biological insights. (B) Single-cell mutational analysis can yield detailed phylogenetic trees to identify which mutations occur in specific unique subclones and which track together. In this example, multiple sCNA and sSNV were interrogated by using targeted sequencing to establish the underlying clonal architecture of the leukemia. This demonstrated convergent loss of TP53 with 17p deletion in 1 subclone and TP53 mutation in another, which was further corroborated by conventional FISH analysis by Wang et al.50
To address this, Zhao et al49 applied single-cell WGS to a total of 116 CLL cells across 2 time points, which were individually isolated into polymerase chain reaction (PCR) tubes and subjected to DNA amplification using multiple displacement amplification. sCNAs were identified at the single-cell level that matched bulk single nucleotide polymorphism analysis, but through single-cell analysis, it was possible to define how sCNAs across the entire genome segregated. They demonstrated that even at the earliest time point, sCNAs were distributed across 5 unique subclones, and after therapy, the emergence of trisomy 12 was restricted to a single subclone.49 However, although single-cell WGS could reliably detect sCNA events, detection of sSNVs was limited because of its generally lower coverage. This study was further limited to a detailed analysis of cells from a single patient, suggesting that strategies to increase throughput (ie, using droplet-based technologies) and to increase resolution will be needed to ensure their greater utility for dissecting clonal architecture in larger numbers of patients.
In contrast to single-cell WGS and/or WES, several studies have evaluated the coexpression of CLL-associated mutated events through targeted assays applied to single CLL cells.50,51 In 1 study, Wang et al50 designed a multiplex PCR assay based on prior bulk sequencing results that analyzed up to 201 amplicons per cell. By using this method, 1152 cells from 5 patients were analyzed, generating precise subclonal architectures based on both sCNA and sSNV aberrations. For example, in 1 patient, 2 distinct subclones resulting in inactivation of TP53 were identified: 1 as a result of a 17p deletion and the other as a result of an inactivating mutation (Figure 3B). This is an example of convergent evolution, which was also seen in other patients and has been described in other cancers.52,53 By generating more complete and accurate phylogenetic trees, the authors were also able to identify subclones lacking clear driver mutations, and they used their approach to validate the impact of novel mutations in WNK1 and LCP1 on cancer-driving functions.50
Methylation, chromatin assembly,54 and histone modification14 are key epigenetic mediators of biology; bulk assessment by dense array technology reveals global hypomethylation with distinct areas of hypermethylation in CLL compared with normal B cells.55-57 Applying reduced representation bisulfite sequencing (RRBS) to CLL revealed high levels of disordered methylation as measured by the proportion of discordant reads (PDR), which itself has been linked to the presence of subclonal driver mutations and adverse outcomes.13 In 1 study, single-cell RRBS demonstrated that CLL cells have more uniform PDR compared with normal B cells that show much greater cell-to-cell variation. Thus, PDR may function as a molecular clock, with uniformly high PDR reflecting multiple divisions of a CLL precursor cell, whereas variable PDR in nonmalignant B cells reflect the admixture of naïve and memory B cells of various ages.58 This group also used single-cell RRBS to reconstruct the phylogenetic relationships between CLL cells. The subsequent lineage trees show early branching in contrast to normal B cells, consistent with rapid evolutionary drift after initial malignant transformation in CLL.59 They also demonstrated linkage between single-cell transcriptional profiles and the underlying methylome, with transcriptional similarity between cells decreasing with increasing phylogenetic distance.59
More recently, Beekman et al14 performed a global assessment of the CLL epigenome, including methylation, chromatin assembly, and histone modification status. This work revealed that CLL cells show similarities in chromatin assembly with multiple stages of differentiating B cells (from naïve to plasma cells), such that unmutated IGHV CLL cells show features of proliferating germinal center (GC) B cells despite their cell-of-origin being pre-GC. In addition, by linking DNA mutations to the epigenome, they demonstrated that mutated MYD88 and trisomy 12 CLL represent distinct molecular subgroups based on chromatin activity and accessibility.14 The application of single-cell techniques such as single-cell assay for transposase-accessible chromatin sequencing (scATAC-seq)60,61 and single-cell chromatin immune-precipitation sequencing62 to further dissect these various facets of CLL can provide additional context to our understanding of the bulk CLL epigenome. For example, scATAC-seq, as applied to acute myeloid leukemia, has uncovered the intriguing expression of multiple, normally distinct regulatory programs in single cells.63
Transcriptomic analysis
Bulk transcriptomic studies using microarrays or via transcriptome sequencing have demonstrated that thousands of genes are differentially expressed between normal B cells and CLL cells. These studies have led to the search for unique transcriptional signatures of CLL as well as discriminating antigens.64,65 For example, 1 study identified that gene expression associated with poor prognosis was enriched for transcripts involved in BCR signaling similar to that seen in CLL lymph nodes, despite cells being isolated from the peripheral blood.29 Furthermore, although both mutated and unmutated IGHV CLL are associated with differing cells of origin,3,4 DNA mutations,66 and epigenetic profiles,56 bulk transcriptional profiles have revealed considerable overlap of these subtypes. Altogether, these collective studies suggest that a single-cell approach may be needed to identify transcriptionally defined subpopulations (Figure 3A).
A range of methodologies has been developed to capture the global transcriptional profiles of single cells, and we direct readers to previous comprehensive reviews22,40,67 for more information. Briefly, techniques that analyze full-length transcripts provide more reliable information about isoforms, splicing events, and SNVs, but they require processing as separate single-cell libraries for a greater number of steps than tag-based methods, which reduces throughput and increases the relative costs. In comparison, tag-based methods incorporate specific oligonucleotides that can include both unique cell barcodes and unique molecular identifiers, which enable increased multiplexing, earlier pooling of libraries, and reduced costs per cell. Furthermore, tag-based methods provide more accurate estimation of transcript abundance because of the inclusion of the unique molecular identifiers, but they sequence only a small portion of each transcript.
Zhao et al49 used a plate-based Smart-Seq2 protocol68,69 that captures full-length transcript data to investigate the transcriptional signatures of 362 CLL cells across 5 time points spanning 18 years and consisting of 35 to 126 cells per time point in a single patient. Through hierarchical clustering and principal component analysis, cells could be divided into 6 unique clusters, which varied over time and therapy. Interestingly, the predominant cluster at the earliest time point, which was enriched for MAPK4, ERBB4, and PDGFRA1 transcripts, diminished with disease progression but re-expanded when the patient was in a partial remission after a splenectomy 16 years later. A separate study that assessed global transcriptional profiles of 289 single CLL cells from 4 patients identified differences in biological processes, including cell cycle and immune responses, which corroborated corresponding DNA mutational signatures and also identified deregulation of genes involved with phospholipid binding and protein folding, facets of CLL biology that were previously unappreciated.50
As an adjunct to transcriptome-wide profiling at the single-cell level, targeted single-cell reverse transcription PCR–based analysis can provide consistent information about transcripts that is not reliably captured by whole transcriptome studies and can also enable increased throughput. Wang et al44 demonstrated the ability to detect mutations in SF3B1 in single cells and also used primers for genes known to be differentially spliced as a result of the K700E mutation to confirm aberrant splicing at the single-cell level. This aberrant single-cell splicing was seen for 4 of 5 SF3B1 mutations (E622D, K666Q, K700E, G742D) but not Q903R, which is localized outside the mutation-rich hotspot region of SF3B1. The results highlighted that, although SF3B1 mutations give rise to disordered splicing, the exact gene targets seem to be mutation dependent.
The cellular transcriptional state of CLL cells can also be affected by epigenetic mechanisms. Landau et al13 previously analyzed the relationship between promoter methylation PDR and the subsequent transcriptional profile; high PDR were associated with lower but more variable gene expression. Through single-cell analysis, it was apparent that genes with promoters exhibiting high PDR tended to be expressed in larger numbers of cells at lower levels, whereas genes with promoters exhibiting low PDR tended to be expressed in smaller numbers of cells but at a higher expression magnitude. This analysis thus provided insight into the impact of locally disordered methylation on cell-to-cell transcriptional variability.
An important question in defining the clonal evolution of CLL is whether mutations arise in response to pressure induced by therapy or whether a small clone of resistant cells is present at the outset of the disease and these resistant cells gain a growth advantage in response to therapy. Burger et al12 examined clonal architecture and evolution in response to chemoimmunotherapy and to the irreversible Bruton tyrosine kinase (BTK) inhibitor ibrutinib by assessing single cells at the complementary DNA level. This approach is more sensitive than previous methods that analyzed genomic DNA because there are many more RNA molecules than DNA molecules per cell. In 1 patient, exposure to fludarabine, cyclophosphamide, and rituximab led to the eradication of a major subclone harboring an SF3B1 mutation, whereas ibrutinib therapy at a subsequent relapse led to the emergence and expansion of 4 distinct mutations in the downstream target of BTK, PLCγ2, which is thought to contribute to ibrutinib resistance.70-72 Although bulk sequencing could identify these mutations on treatment, only single-cell sequencing could demonstrate that these 4 mutations were present in mutually exclusive distinct subclones. In addition, a sensitive targeted approach that harnessed high-throughput droplet microfluidics was used to directly identify rare individual cells bearing resistance-associated mutations in pretreatment samples,12 which supported earlier predictions that resistant cells are present before treatment is initiated.73
This approach relied on a two-stage design. In the first stage, individual cells were encapsulated in droplets, allele-specific quantitative PCR was performed with a fluorescent probe in each droplet, and the number of mutant cells was determined by counting the number of fluorescent droplets. This stage achieved detection at a level of 1 in 1000 cells. For the second stage, the allele-specific PCR was performed by using a larger number of encapsulated cells, the emulsion was broken, and the total amount of amplicon was quantified by digital PCR. This quantification was related back to cellular concentration by comparing to a standard curve prepared by spiking a known number of mutant cells into a normal cell background. By using this second approach, the authors were able to detect a PLCG2-M1141R mutation that was present before ibrutinib was initiated at a frequency of 1 in 500 000 cells (0.0002%), which confirmed the hypothesis that a minuscule number of resistant clones are present at the outset of the disease. These data highlight the evolutionary capacity of leukemia cells and provide an understanding of the mechanics that underlie the kinetics of relapse after the strong selective pressure imposed by targeted inhibitor therapy.
Using single-cell RNA sequencing to study a range of physiological and pathological disease processes within hematology has provided a better understanding of normal hematopoiesis25 as well as better characterization of acute myeloid leukemia and chronic myeloid leukemia stem cells,74,75 circulating and relapsed plasma cells,76,77 megakaryocyte differentiation in myelofibrosis78 and therapy-resistant minimal residual disease–positive acute lymphoblastic leukemia cells.79 Furthermore, a recent study by Milpied et al80 characterized GC B cells at single-cell resolution, integrating phenotypic, genetic, and transcriptomic analyses to demonstrate that follicular lymphoma B cells are not transcriptionally arrested in a GC B-cell state but have their own unique transcriptional signature. Similar investigations of CLL, which build upon the insights made thus far (summarized in Table 1), will undoubtedly reveal nuances of CLL biology in its natural environment.
Investigating accessory cells in CLL
It has long been acknowledged that CLL (and indeed the other B-cell malignancies) have close interactions with the microenvironment. Given the heterogeneity of immune cell populations, single-cell analysis provides advantages over conventional bulk approaches and is highly relevant because the tumor microenvironment is critical to the pathogenesis of CLL and other lymphoproliferative disorders. For example, bulk gene expression studies of follicular lymphoma demonstrate that the gene signature of the infiltrating immune cells is a key determinant of outcomes.81 In addition to this, we are in the midst of an immunotherapy revolution with the increasing use of checkpoint inhibition,82 chimeric antigen receptor (CAR) T-cell therapy,83,84 bispecific T-cell engaging antibodies,85 and cancer vaccines.86 Information from these immunotherapy studies is highly relevant for CLL because CLL is characterized by T-cell dysfunction, increased levels of exhaustion markers, abnormal immunologic synapse formation, reduced proliferation, and muted cytotoxicity.87-89 Indeed, insights to be gained from single-cell analysis have the potential to shed light on the basis of the poor responses seen in CLL in response to checkpoint inhibitors82 and lower responses to CAR T-cell therapy compared with acute lymphoblastic leukemia.90 A better understanding of the factors that modulate the immune system at a single-cell level is therefore timely for developing rational therapeutics and synergistic combinational strategies.
Flow cytometry has been used extensively over the years for characterizing the immune landscape in CLL as well as defining their functional states. For example, these studies have revealed CLL to have increased circulating FoxP3+ T regulatory cells,91 skewed proportion of Th17 cells,92 dysregulated natural killer cells,93 and increased levels of nonclassical monocytes.94 More recently, ibrutinib has been shown to reverse these defects, which allows enhanced function of CAR T cells84,95-97 and bispecific antibodies,85 whereas immunomodulation with lenalidomide allows recovery of synapse formation.88 Although flow cytometry is an extremely useful and high-throughput technique, it is limited by the number of antigens that can be simultaneously resolved because of the spectral overlap of fluorophores. Mass cytometry is a higher-dimensional approach that uses antibodies conjugated to metal isotypes (in contrast to fluorophores) followed by sample ionization and time-of-flight mass spectrometric analysis. Metal isotopes overcome limitations with spectral overlap so that increased numbers of antigens can be resolved simultaneously, but unlike flow cytometry, cells cannot be recovered for downstream functional analysis. Mass cytometry has been useful in higher-order characterization of immune cell subsets in a range of malignancies including Hodgkin lymphoma98 and identification of disease-associated immune phenotypes that correlate with response rates.99 This technology has become an established technique in its own right. Within CLL, initial data reveal highly variable expression of inhibitory receptors between patients,100 but T cells overall express multiple checkpoint molecules and these molecules are at higher levels compared with normal controls. In addition, CD8+ T cells demonstrate a predominantly senescent phenotype.101 Another approach to analyzing tumor-associated immune cells is with single-cell RNA (scRNA) sequencing in parallel with tumor assessment. This overcomes the limitations of prior selection of antibodies for flow or mass cytometry, which allows unbiased and higher-complexity analysis. In breast cancer, it has provided a compendium of infiltrating immune cells and identified a CD8+ tissue-resident memory signature associated with enhanced survival.102,103 By focusing on individual T cells, it has linked T-cell receptor clonotypes with their underlying cellular activation state. Assessment of T cells within the CLL microenvironment, given their potential for antileukemic effects and inherent dysfunction, are therefore of great interest. In addition to the immune component, stromal cells provide survival factors, chemokines, cytokines, and cell surface proteins such as integrins that enhance CLL survival. Although much progress has been made in understanding their contribution to CLL biology, further mechanistic insights into how stromal cells support CLL can be obtained through single-cell analysis. scRNA sequencing has led to an appreciation of the complexity of lymph node stromal cells (albeit in mice, but with likely parallels in humans)104 as well as the supportive relationship between stromal cells and tumor cells in solid tumors.105,106
Multiomic contextual assessment
As single modality assays have become increasingly available, newer assays that aim to capture multidimensional genomic, transcriptomic, and protein information from a single cell have been emerging.107-109 For example, cellular indexing of transcriptomes and epitopes by sequencing (CITE-seq) uses oligonucleotide-tagged antibodies to assess protein expression in combination with scRNA sequencing, thereby providing phenotype-transcriptional correlation.39 This approach overcomes the limitation that single-cell transcriptome analysis alone cannot consistently detect the expression of surface markers used for classifying cell subsets.39 These efforts promise to provide a more complete insight by linking genetic aberrations with transcriptional signatures and/or protein expression,110 with the aim of combining all of these assays into a single platform.
Despite advances in technologic assays looking at global or targeted gene expression or circulating free DNA, immunohistochemistry is still central to diagnosis, because it provides useful information about the overall architecture, cell types, and level of infiltration within biopsies and resection samples. Combining these types of data with single-cell approaches could be highly informative by enabling correlation of cell-cell interactions within the microenvironment. This would show us how adjacent cells interact and define their consequent transcriptional and proteomic profiles. In addition, single-cell analysis could identify rare cell populations of interest, and correlative studies of their location would help assess their functional impact. One example of this type of endeavor is imaging mass cytometry in which tissue sections are stained with metal isotope-labeled antibodies, and protein expression is linked to location by using mass cytometry analysis.111 An alternative is the use of single-molecule RNA FISH, which uses short conjugated probes to determine mRNA expression and spatial distribution within cells and tissue sections.112 Within CLL, this has clear applicability for assessing the lymph node and marrow microenvironment, especially within pseudofollicles, which represent a unique site of CLL proliferation and where there is also a substantial presence of T cells.
Clinical applications of insights from single-cell analysis
With the vast amount of information that will be generated from ongoing and future single-cell studies in CLL and across hematologic disorders overall, we can anticipate relating this new knowledge back to the patient. Advances in bulk genomics have defined the spectrum of mutations and genomic changes within CLL and are undergoing evaluation for impact on prognosis and prediction of outcomes in CLL,11,72,113-115 as well as incorporation of newer epigenetic and transcriptional insights.116-119 Single-cell level information can provide valuable insights for CLL patients at diverse points in their disease course (Figure 4). At diagnosis, when leukemia cells are in abundance, single-cell analytics will enable a detailed snapshot of the precise composition of the leukemia and also an assessment of the accessory immune cells. By informing us about the classification of the patient’s CLL into a particular biologic subgroup and/or the status the patient’s host immune microenvironment, we will be better able to provide advanced prognostication beyond what is possible with traditional cytogenetics and global mutational profiles. Consequently, more accurate decisions on the timing and modality of therapy can be made. For example, when therapy is initiated, assessment of mutational status of key genes or distinct transcriptional profiles implicated in resistance would help optimize treatment decisions, especially in an era in which many targeted agents are available. Indeed, identifying multiple subclones may highlight a subclone associated with higher risk and warrant subclone-specific therapy. In addition, coevaluation of the immune system will help identify the role of the many potential novel immunomodulatory agents on a patient-specific basis. This will maximize targeted efficacy through rational deployment, such as combinational use of checkpoint inhibitors beyond CTLA-4 and the PD-1/PD-L1 axis, reversal of inhibitory pathways limiting T-cell responses, and better targeting of microenvironmental support. After initial therapy, single-cell technology provides the sensitivity needed to enable screening for the emergence of resistant clones, which would potentially allow tailored treatment early in the course of relapse and minimize ongoing clonal evolution associated with treatment resistance. An ever-present clinical consideration in CLL is the potential to undergo Richter’s transformation, for which outcomes are poor and treatments are limited, especially in the context of advanced age. Single-cell approaches to understanding this disease entity are therefore urgently required to help better understand the disease process and enable development of therapeutics. Here again, one can certainly envision a scenario in which monitoring the peripheral blood may detect a Richter’s signature and lead to earlier detection, treatment optimization, and improved outcomes for patients. The high costs associated with scRNA sequencing combined with the requirement for dedicated bioinformatics workflows to obtain meaningful interpretation has to be balanced against the cost of novel cancer therapies. Potential savings are possible by choosing the optimal therapy at the outset and by sensitive monitoring to detect resistance to treatment early, which will allow prompt discontinuation of treatment and appropriate re-treatment. With increasing knowledge of single-cell analytics, one can envisage this technology being incorporated into clinical trial design to determine its wider applicability.
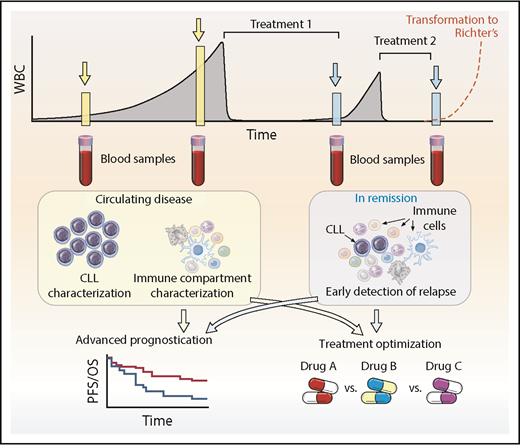
Clinical application of single-cell technologies for CLL. Single-cell approaches allow for deeper characterization of leukemic and immune cells to help aid prognostication, optimize and rationalize treatments, and modulate the immune system to enhance potential immunotherapeutic approaches at diagnosis and relapse. At diagnosis or during follow-up, monitoring can identify clones that represent higher risk, and appropriate treatment can be instituted or defects within immune cells can be characterized and then addressed. After treatment, single-cell approaches have the potential to detect relapse earlier, identify the underlying resistance-driving mutations, and select the optimum strategy for overcoming the problem. An ever-present clinical concern is Richter’s transformation, in which single-cell approaches may also be able to help with earlier diagnosis and treatment optimization. This approach is not limited to CLL but has applicability across the range of hematologic malignancies. PFS, progression-free survival; OS, overall survival; WBC, white blood cell.
Clinical application of single-cell technologies for CLL. Single-cell approaches allow for deeper characterization of leukemic and immune cells to help aid prognostication, optimize and rationalize treatments, and modulate the immune system to enhance potential immunotherapeutic approaches at diagnosis and relapse. At diagnosis or during follow-up, monitoring can identify clones that represent higher risk, and appropriate treatment can be instituted or defects within immune cells can be characterized and then addressed. After treatment, single-cell approaches have the potential to detect relapse earlier, identify the underlying resistance-driving mutations, and select the optimum strategy for overcoming the problem. An ever-present clinical concern is Richter’s transformation, in which single-cell approaches may also be able to help with earlier diagnosis and treatment optimization. This approach is not limited to CLL but has applicability across the range of hematologic malignancies. PFS, progression-free survival; OS, overall survival; WBC, white blood cell.
Although the field of single-cell technology has rapidly evolved over the last few years and continues to advance rapidly, the recent emergence of high-throughput relatively user-friendly methods means that high levels of information can readily be gleaned from single leukemic or supporting cells, which makes it feasible to thoroughly investigate all aspects of cellular biology across the disease course of CLL. Using gene editing technologies to enable paired assessment of single-cell gene perturbation combined with scRNA sequencing will provide additional insights.120,121 Assuredly, the ability to analyze thousands of cells in multiple patients and ongoing work in this area, akin to the advances with bulk sequencing, will catalyze our understanding of CLL biology with unparalleled detail. We expect that these insights will help transform our knowledge of the pathophysiology and clinical behavior of CLL, help guide the selection of optimal therapeutics, and enable appropriate monitoring, with the aim of improving patient outcomes.
Acknowledgments
The authors thank Ken Livak for his critical reading of the manuscript.
This work was supported by grants from the Wellcome Trust (WT101658AIA) and the Kay Kendall Leukaemia Fund (KKL1215) (S.H.G.), and from the National Institutes of Health, National Cancer Institute (1R01CA182461-02, 1R01CA184922-01, U10CA180861-01, 1RO1CA155010-02) (C.J.W.). C.J.W. is a Scholar of the Leukemia and Lymphoma Society.
Authorship
Contribution: S.H.G. and C.J.W. wrote the paper.
Conflict-of-interest disclosure: C.J.W. is a cofounder of Neon Therapeutics and is a member of its scientific advisory board. S.H.G. declares no competing financial interests.
Correspondence: Catherine J. Wu, Department of Medical Oncology, Dana-Farber Cancer Institute, Dana 520, 44 Binney St, Boston, MA, 02115; e-mail: cwu@partners.org
