

Abstract
The classical model of hematopoiesis proposes a hierarchy in which a small number of multipotent hematopoietic stem cells (HSCs) maintain all blood lineages by giving rise to progeny that pass through discrete progenitor stages. At each stage, lineage differentiation potential is restricted, coupled with the loss of ability to self-renew. Recently, single-cell approaches have been used to test certain assumptions made by this model, in particular relating to megakaryocyte (Mk) and erythroid (E) development. An alternative model has emerged in which substantial heterogeneity and lineage-priming exists within the HSC compartment, including the existence of multipotent but megakaryocyte/platelet-biased HSCs. Hematopoietic differentiation follows a hierarchical continuum, passing through cellular nodes and branch points. Megakaryocytes are produced via a shared pathway with the erythroid lineage, also shared in its early stages with mast cells, eosinophils, and basophils, but separate from other myeloid and lymphoid lineages. In addition, distinct pathways for direct differentiation of Mk from HSCs may coexist and could be important in situations of increased physiological requirements or in malignancies. Further work at single-cell resolution using multiomic approaches and examining Mk-E biased subsets within their physiological context will undoubtedly improve our understanding of normal hematopoiesis and ability to manipulate this in pathology.
Introduction
Hematopoietic stem cells (HSCs) were discovered half a century ago when the observation was made that transplanting adult bone marrow cells could regenerate multiple blood cell types after radiation injury.1-3 Methods for isolating populations of hematopoietic stem/progenitor cells (HSPCs) were subsequently pioneered4,5 and led to the concept of a hematopoietic hierarchy to explain how rare, self-renewing, multipotent cells were able to generate and maintain mature blood cells throughout life and during periods of physiological stress.6 HSCs were proposed to differentiate via stepwise transitions through discrete oligo- and unipotent stages, with loss of multipotency coupled with loss of self-renewal capacity. This model became a paradigm for other tissue-specific stem cells. The first lineage bifurcation downstream of HSC and multipotent progenitors (MPPs) was considered to be between progenitor cells that contained all myeloid (common myeloid progenitors; CMPs4 ) or all lymphoid potentials (common lymphoid progenitors7 ). In megakaryopoiesis and erythropoiesis, CMPs were proposed to give rise to a population of bipotent megakaryocyte (Mk)-erythroid (E) progenitors (MEPs),4,5,8,9 with other myeloid lineages encompassed within granulocyte-macrophage progenitors (GMPs). These oligopotent progenitors generated unipotent progenitors and mature cell types (Figure 1).
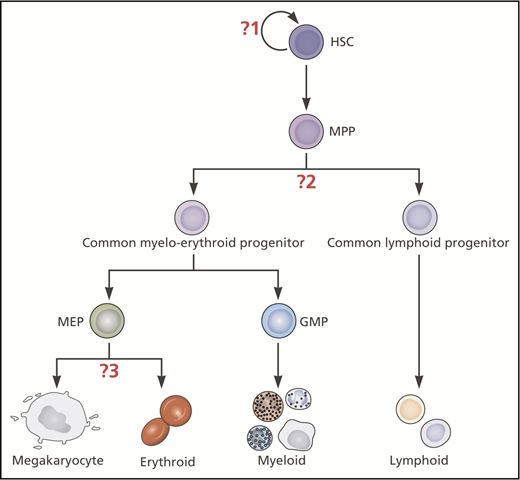
Classical hierarchical model of hematopoiesis illustrating where insights from single-cell studies have challenged 3 key assumptions. The question marks indicate the 3 key assumptions in this model that have been challenged by recent insights from single-cell studies: (1) Mk-E cells are generated by a homogeneous population of multipotent, self-renewing HSCs. (2) The first lineage bifurcation separates progenitors with Mk-E/myeloid from those with lymphoid capacity. (3) Mk and E potentials are closely affiliated through to late stages of hematopoietic development. Myeloid and lymphoid subsets exist but are not shown in this figure. Professional illustration by Patrick Lane, ScEYEnce Studios.
Classical hierarchical model of hematopoiesis illustrating where insights from single-cell studies have challenged 3 key assumptions. The question marks indicate the 3 key assumptions in this model that have been challenged by recent insights from single-cell studies: (1) Mk-E cells are generated by a homogeneous population of multipotent, self-renewing HSCs. (2) The first lineage bifurcation separates progenitors with Mk-E/myeloid from those with lymphoid capacity. (3) Mk and E potentials are closely affiliated through to late stages of hematopoietic development. Myeloid and lymphoid subsets exist but are not shown in this figure. Professional illustration by Patrick Lane, ScEYEnce Studios.
This hierarchical model was largely based on experiments with populations of cells that were initially considered to be homogeneous. Studies using paired daughter cell assays, single-cell transplants, and other approaches have, over many years, demonstrated significant heterogeneity among HSPC populations.10,11 The recent explosion in single-cell omics (eg, genomics, transcriptomics, epigenomics, proteomics, metabolomics)12 has enabled a much finer dissection of cellular heterogeneity than was previously possible.13-15 Substantial heterogeneity has been uncovered within previously defined HSPC populations. With the advent of high-throughput platforms for simultaneously profiling tens of thousands of individual cells, novel, rare subpopulations have been described and cells ordered over pseudotime to suggest differentiation trajectories, although very few studies (if any) have actually proven a pseudotime journey.16-18 The resulting insights have definitively demonstrated that analysis of HSPCs at the population level masks extensive heterogeneity of lineage potential and bias, and that priming toward specific differentiation pathways is present from the earliest HSCs.19-21
The vast majority of cells produced by the bone marrow are of Mk/platelet and E lineages. Defining the distinct HSPC subsets with Mk and E potential, their hierarchical and lineage relationships and branch points are of crucial importance for regenerative medicine and understanding perturbations of hematopoiesis in disease. In this review, we focus on how single-cell approaches have been used to test certain assumptions relating to Mk-E development in the classical model of hematopoietic development (Figure 1): (1) Mk-E cells are generated by a homogeneous population of multipotent, self-renewing HSCs; (2) the first lineage bifurcation separates progenitors with Mk-E/myeloid from those with lymphoid capacity; and (3) Mk and E potentials are closely affiliated through to late stages of hematopoietic development.
Key concepts and experimental challenges in the study of Mk-E development
Given the ready accessibility of the cells and well-developed experimental approaches, hematopoiesis is one of the most thoroughly studied cellular systems. However, all experimental assays are associated with certain limitations, and there are several key challenges that are specific to studying Mk-E development that underlie many of the conceptual challenges discussed in this review. Terminology used in the field can also sometimes be overlapping (see Table 1).
Unique and overlapping commonly used stem cell terms and definitions
| Term | Widely accepted definition |
|---|---|
| Long-term HSC | Cells able to reconstitute all 5 types of mature blood cells in recipient mice over the long term (≥16-wk after transplantation) and in secondary and tertiary transplants28 |
| Intermediate-term repopulating HSC | Cells that reconstitute multilineage blood cells over the medium term (6-8 mo), but show some loss of self-renewal in secondary transplant recipients28 |
| Short-term repopulating HSC | Cells that transiently reconstitute multiple blood cell types for up to 8 wk in transplant recipients, or in which 1 or more donor-derived lineages disappear before 24 wk after primary transplant |
| Lineage priming | Expression of a transcriptional program associated with potential for differentiation to mature cells of a specific lineage in a multi/oligopotent cell |
| Lineage bias | Multi-/oligopotent cells that preferentially give rise to a single lineage but retain potential for alternative differentiation |
| Lineage restricted/committed | Oligo-/unipotent cells not able to give rise to cells of certain lineages |
| Lineage potential | The mature lineages that a cell has the potential to give rise to, depending on external stimuli |
| Lineage fate | The lineage that a stem/progenitor will give rise to in vivo |
| Term | Widely accepted definition |
|---|---|
| Long-term HSC | Cells able to reconstitute all 5 types of mature blood cells in recipient mice over the long term (≥16-wk after transplantation) and in secondary and tertiary transplants28 |
| Intermediate-term repopulating HSC | Cells that reconstitute multilineage blood cells over the medium term (6-8 mo), but show some loss of self-renewal in secondary transplant recipients28 |
| Short-term repopulating HSC | Cells that transiently reconstitute multiple blood cell types for up to 8 wk in transplant recipients, or in which 1 or more donor-derived lineages disappear before 24 wk after primary transplant |
| Lineage priming | Expression of a transcriptional program associated with potential for differentiation to mature cells of a specific lineage in a multi/oligopotent cell |
| Lineage bias | Multi-/oligopotent cells that preferentially give rise to a single lineage but retain potential for alternative differentiation |
| Lineage restricted/committed | Oligo-/unipotent cells not able to give rise to cells of certain lineages |
| Lineage potential | The mature lineages that a cell has the potential to give rise to, depending on external stimuli |
| Lineage fate | The lineage that a stem/progenitor will give rise to in vivo |
The fundamental limitation is that cells may behave differently, depending on the experimental system employed and whether it is testing differentiation potential, self-renewal, lineage bias, or unperturbed fate. This particularly becomes an issue when dealing with assays of single cells, in which only a single assay is possible for any single cell. Therefore, the absence of a particular lineage read-out in any specific assay cannot correctly be interpreted as a lack of potential for differentiation to that lineage in all conditions (Figure 2). Cells may simply behave differently depending on external stimuli or other components of the experimental system employed.
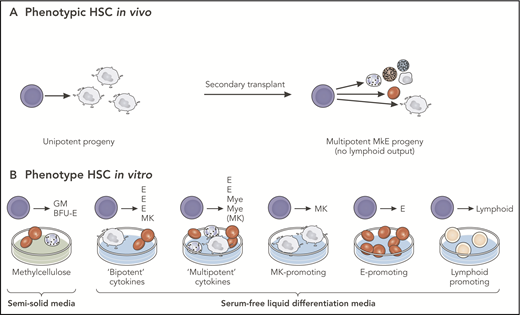
Observed vs potential differentiation of multipotent progenitors. Cells behave differently, depending on the experimental system employed. The absence of a particular lineage read-out in any specific assay cannot correctly be interpreted as a lack of potential for differentiation to that lineage in all conditions. (A) Analysis of a single-phenotypic HSC in vivo that may appear to be unipotent in primary transplants may reveal multipotent differentiation capacity in secondary transplants. (B) Analysis of progeny of the same HSC in vitro may demonstrate differentiation into single- or multiple-cell types, depending on the culture conditions. In this example, the single HSC studied is multipotent, a conclusion that can only be reached by multiple different lineage potential assays in combination. BFU-E, blast forming unit-erythroid; GM, granulocyte-monocyte; E, erythroid; MkE, megakaryocyte-erythroid; Mye, myeloid. Professional illustration by Patrick Lane, ScEYEnce Studios.
Observed vs potential differentiation of multipotent progenitors. Cells behave differently, depending on the experimental system employed. The absence of a particular lineage read-out in any specific assay cannot correctly be interpreted as a lack of potential for differentiation to that lineage in all conditions. (A) Analysis of a single-phenotypic HSC in vivo that may appear to be unipotent in primary transplants may reveal multipotent differentiation capacity in secondary transplants. (B) Analysis of progeny of the same HSC in vitro may demonstrate differentiation into single- or multiple-cell types, depending on the culture conditions. In this example, the single HSC studied is multipotent, a conclusion that can only be reached by multiple different lineage potential assays in combination. BFU-E, blast forming unit-erythroid; GM, granulocyte-monocyte; E, erythroid; MkE, megakaryocyte-erythroid; Mye, myeloid. Professional illustration by Patrick Lane, ScEYEnce Studios.
In vitro assays of lineage potential are frequently used but are inherently biased by selection of cytokines, culture medium and conditions, and assay times. This is a particular issue for Mk-E lineages, which differ markedly in many aspects, including rates of cell division. Limited assays are available to test Mk-E bi-potency, particularly for human cells. Liquid culture systems can easily be biased toward detection of E progeny, as E progenitors vastly outnumber Mk, especially at later times. Methylcellulose colony-forming assays are not conducive to Mk growth, and Mk-specific collagen-based semisolid colony-forming assays (eg, Megacult) have low clonogenicity precluding robust single-cell assays, although they can be adapted to give rise to both E and Mk with the addition of erythropoietin,22,23 and a plasma clot assay was similarly also used previously to support both Mk and E colony growth.24
In vivo assays also have challenges and limitations, particularly for the assessment of human cells. Irradiation and transplantation do not assess homeostatic hematopoiesis, and as mature platelets and red blood cells do not express CD45, using the CD45.1/CD45.2 alloantigen in vivo tracking system is not possible. Further, fluorescence-activated cell sorting (FACS) isolation of mature Mks is very challenging, so studies are usually limited to platelets or Mk progenitors. In vivo lineage tracing can also be employed. However, as red blood cells and platelets lack DNA, DNA barcoding can only be used to study their progenitors, and this may introduce certain biases. Inducible genetic recombination lineage tracing models25 (eg, platelet factor 4-Cre26 ) can also be used, but often lack fidelity with expression of the Cre in HSPCs and other lineages.
Taking these limitations into account is of crucial importance when considering the cellular pathways of Mk-E specification discussed in the following sections.
Heterogeneity of lineage fate among multipotent HSCs
The classical model of hematopoiesis proposed that all blood lineages including Mk-E are continuously replenished by multipotent HSCs. However, many HSCs show differentiation bias, preferentially giving rise to specific lineages. The first distinct HSC subsets to be identified were those stably biased toward lymphoid or myeloid output,27,28 although the experimental systems used in these experiments did not allow the study of Mk-E differentiation. In paired daughter cell assays in vitro, and in primary and secondary single cell transplants in vivo, asymmetrical HSC divisions were observed that resulted in 1 HSC plus a Mk or myeloid restricted progenitor.29 Ten percent of murine long-term (LT)-HSC expressed the Mk surface antigen CD41 and contained self-renewing cells with 3 patterns of restricted differentiation output (common myeloid-like [20%], Mk [12%], and Mk-E [2%]), as well as intermediate and short-term (ST)-HSCs.29 This seminal work indicated that lineage restricted progenitors could have long-term repopulating activity, challenging the prior assumption that only multipotent HSCs at the top of the differentiation hierarchy could self-renew.29
Further evidence for Mk/platelet primed HSCs was reported simultaneously by 2 groups. First, c-Kitlo murine HSCs were found to be enriched in quiescent LT-HSCs, whereas c-Kithi cells were Mk biased.30 Second, expression of the megakaryocyte lineage marker von Willebrand Factor (VWF) in long-term repopulating and self-renewing stem cells had been previously demonstrated in key papers establishing the immunophenotypic identify of LT-HSC in mice.31,32 Subsequently, studies using a transgenic (VWF)–green fluorescent protein mouse model33 demonstrated that around half of murine LT-HSCs expressed the Mk gene VWF and gave platelet-biased reconstitution with considerable myeloid but low lymphoid contribution.33 Single VWF+ HSCs coexpressed Mk, E, and myeloid- but not lymphoid-affiliated genes. After acute platelet depletion, VWF+ HSCs were selectively recruited from quiescence to active cell cycling, rapidly replenishing circulating platelets. Almost all embryonic fetal liver HSCs were found to be VWF+,15 and these VWF+ HSCs reconstituted both VWF+ and VWF− HSCs with maintained differentiation bias in serial transplants, whereas VWF− HSCs were unable to give rise to VWF+ HSCs. Together, these experiments indicate that platelet-primed HSCs developmentally and hierarchically precede lymphoid-biased (VWF−) HSCs.33
There has been some debate in the literature about whether cells in the HSC compartment are Mk-biased vs committed/restricted (Table 1). In addition to reports of Mk-biased HSCs, separate studies have described unipotent Mk progenitors arising directly from HSCs in mice, suggesting that cells can bypass intermediate progenitor steps, as described in the classical hierarchy. For example, direct differentiation of immunophenotypic LT-HSCs into Mks was observed using live-cell imaging, in some cases occurring without cell division.34 However, as discussed earlier, the observation that a cell gives rise to only 1 lineage in a particular assay does not necessarily imply that this cell did not have multipotent potential at the outset of the experiment, but may reflect limitations and/or biases in the experimental approach or the presence of contaminating unilineage progenitors among the LT-HSCs isolated for study (Figure 2).
Whether or not Mk-biased HSCs retain multipotency was recently definitively addressed. In more than 1000 single-cell HSC transplants, just more than 10% of VWF+ LT-HSCs stably replenished Mk/platelets but not other lineages, whereas ∼90% replenished other lineages in addition to Mk/platelets. Crucially, Mk-biased HSCs showing lineage restriction in primary transplants consistently sustained multipotency in vitro and in secondary transplants.35 The explanation as to why apparently Mk-restricted HSCs give rise to other lineages when assayed in vitro are not clear. This may reflect either differences in external stimuli or, alternatively, limitations in detection of certain lineages in vivo in the experimental system employed. However, similar findings have been reported for myeloid-biased HSCs that exclusively generated myeloid progeny in primary animals but gave rise to multilineage reconstitution after secondary transplant.35 Distinct patterns of lineage restriction were observed with Mk, Mk-E, Mk-E-myeloid primed, and fully multipotent Mk-E-myeloid-lymphoid HSC subsets. Platelets were the only lineage that was invariably reconstituted in 100% of transplants, followed by E in ∼90%.35 Intriguingly, no HSCs contributed exclusively to any other single blood cell lineage other than platelets, suggesting that HSC priming might be exclusive to the Mk lineage.6
Genetic lineage tracing and transposon tagging experiments have also provided evidence for Mk-biased HSCs in unperturbed murine hematopoiesis.37 In these experiments, although nascent mature cells initially contained unique tags suggesting replenishment by unilineage progenitors, after 4 weeks, significant numbers of shared tags were observed across lineages. Notably, almost half of E clones shared transposon tags with myeloid cells, whereas very few Mk clones were shared exclusively with E cells, as would have been predicted if a shared MEP were the predominant differentiation route in unperturbed hematopoiesis. This suggests a shared origin for E and myeloid lineages, but a largely separate differentiation pathway for Mk in murine hematopoiesis. However, there are some limitations to this approach, including the lack of ability to detect transient states and smaller sized clones, restriction to study of Mk/E progenitors rather than mature platelets/red blood cells, and issues relating to purification strategies for the precursor cells studied.
Diverging paths within MPP: the earliest lineage bifurcation separates Mk-E from lymphoid trajectory
The classical model of hematopoiesis proposed that the first lineage bifurcation separated progenitors with myeloid vs lymphoid potential. Using single-cell assays, it was subsequently demonstrated that the earliest bifurcation occurred in MPPs, immediately downstream of multipotent HSCs, and separated cells with lympho-myeloid but no Mk-E potential from cells with combined Mk-E and myeloid potential (Figure 1). Murine Lin−Sca1+c-kit+ (LSK) cells expressing high levels of the tyrosine kinase receptor Flt3 had granulocyte, monocyte, and B/T lymphoid potential but had lost the ability to sustain Mk or E progeny in vitro and after transplantation in vivo.38 Single-cell molecular profiling confirmed that cells with lympho-myeloid but no Mk-E potential downregulate Mk-E and upregulate lymphoid-associated genes while sustaining myeloid transcriptional programs, whereas Mk-E progenitors lose expression of lymphoid genes,32 highlighting an apparent antagonism between Mk-E and lymphoid fate. Recent work combining in vitro differentiation, index sorting, and single-cell RNA sequencing (capturing the fluorescence intensity of each individual cell during FACS isolation so that transcriptome analysis can be correlated with immunophenotype) on individual human umbilical cord blood HSC/MPP cells and enriched CD49f+ LT-HSCs indicated that the Mk-E/myeloid and lympho-myeloid divergence is initiated in the phenotypic LT-HSC compartment and not at MPP stage in humans.39 Of the multipotent colonies arising in an MS5 stromal cell-based system, around half were multipotent and half unipotent progeny, with very few (<1%) Mk only, which may be a result of the culture conditions being unsupportive for MK differentiation.39
Close affiliation between E and eosinophil/basophil/mast cell lineages
Unique paths for distinct granulocyte subpopulations were demonstrated in experiments using mice expressing green fluorescent protein–tagged GATA1. GATA1+ myeloid progenitors gave rise to Mk-E and eosinophil and mast cells, whereas GATA1− progenitors generated monocytes and neutrophils.40 Importantly, this work challenged the existence of common pre-GM and GMP populations and highlighted a closer link between Mk-E-eosinophil-basophil-mast cell fates than had previously been appreciated.40 This observation was recently extended in a comprehensive single-cell study of Kit+ murine HSPCs focusing on E differentiation.21 Here, reconstructing differentiation trajectories from single-cell RNAseq data indicated coupling of E and basophilic lineages, with earlier divergence of Mk cells from multipotent progenitors, whereas lympho-myeloid differentiation occurred along a separate trajectory.21 Notably, the majority of E-lineage genes were only induced at the onset of E commitment, and not in earlier progenitors.21
Taken together, these data confirm that although the HSC compartment may contain lineage-committed Mk progenitors that are immunophenotypically indistinguishable from nonrestricted progenitors (eg, as observed in live imaging studies34 ), this does not contradict the substantial evidence indicating that Mk-biased self-renewing LT-HSCs are multipotent and capable of regenerating multiple blood lineages, and that a hierarchical model of differentiation with early divergence of lymphoid and close association of Mk-E/eosinophil/basophil/mast cell differentiation trajectories exists, at least in early stages of hematopoiesis. There is also the possibility of an additional developmental pathway for Mk alone arising directly from HSC; however, this remains poorly characterized at the present time.
The physiological and evolutionary relevance of the Mk program in HSCs remains uncertain. A developmental link between Mk and E may have been an important evolutionary pressure to survive challenges such as acute blood loss and chronic anemia to enable rapid and independent generation of Mk or E cells from multipotent cells. It is perhaps surprising that E-biased HSCs have not been identified. Although E specification may be a relatively late event during steady-state hematopoiesis, erythropoietin (EPO) increases in physiological emergencies and EPO-exposed HSC/MPPs give rise to E-biased lineage output after transplantation, indicating that E-directed cell fate programming can be initiated within primitive multipotent cells.41 Similarly, platelet-biased HSCs can rapidly produce platelets in the setting of acute thrombocytopenia.42 Whether similar emergency pathways are required for rapid replenishment of specific lineages (eg, myeloid and lymphoid) remains unclear. However, determining whether permissive or instructive signals direct multipotent progenitors to produce cells of certain lineages in disease remains a challenging and yet crucial question to address.43
Do Mk and E differentiation trajectories remain closely affiliated through to later stages of hematopoiesis?
The cloning of thrombopoietin, the major factor promoting Mk and platelet biogenesis, in 1994 set the stage for in-depth explorations of megakaryopoiesis.44-47 Before this, a close relationship between the E and Mk lineages had been recognized from observations that erythroleukemia cell lines and blasts isolated from patients with biphenotypic leukemias can possess features of both E and Mk lineages, whereas E or Mk with B or T lymphoid characteristics are almost never seen.48,49 In addition, treatment with recombinant EPO can stimulate platelet production in addition to erythropoiesis in both mice and humans.50 Subsequent experiments indicated that exposing CD34+ cells to both EPO and thrombopoietin increased E and Mk progenitors contained within the human CD34+ CD45RA-negative cell fraction.24
In mice, Mk-E progenitors are alternatively referred to as MEP and preMegE, which are identified using different immunophenotypic markers but broadly capture a similar population of cells. Akashi et al divided Lin−Sca1−cKit+ LK progenitors (downstream of LSK, Lin−Sca1+cKit+) into CMP (CD34+FcgRlow), GMP (CD34+FcgRhigh), and MEP (CD34−FcgRlow).4 Multipotent clonogenic assays showed that MEP cells gave rise to Mk-only, E-only, and E + Mk mixed colonies in roughly equal proportions, and suggested a hierarchical relationship in which GMP and MEP arose from CMP.4 Numerous publications later demonstrated significant heterogeneity and lineage specification among both CMP and MEP and proposed an alternative strategy that suggested division of LK into 6 distinct populations including PreMegE and Pre-GM, as well as lineage-committed progenitors. In contrast to Akashi’s data on colony formation in methylcellulose, in which approximately 30% of MEP gave rise to mixed Mk-E progeny in Agar semisolid cultures, less than 10% of PreMegE colonies were mixed Mk/E, with approximately 50% of PreMegE, cells giving rise to Mk only and approximately 50% to E only. Contemporaneous studies confirmed that the majority of individual cells from putative MEP subsets give rise to single lineage progeny, whereas only a minority generate mixed Mk/E colonies, and presumably constitute a bipotent progenitor.8,51
Support for a common developmental pathway between E and Mk lineages also came from observations of shared gene expression and gene regulatory networks; for example, dependence on GATA1, GATA2, FOG1, TAL1, and NFE2 transcription factors and simultaneous defects in both erythropoiesis and megakaryopoiesis in mouse models and patients with defects in these genes.52 Further, antagonistic expression of these transcription factors with others (eg, GATA1 repression of PU.1-dependent transcription) has been shown to direct lineage specification by promoting Mk-E and repressing myeloid programs.53 In evaluating these data, it is important to consider that apparent shared pathways may result from an inability to distinguish cells immunophenotypically and by molecular profiling. Just as populations that are immunophenotypically indistinct should not be assumed to be homogeneous, concordant expression of regulatory molecules (eg, GATA1 in E and Mk differentiation) does not necessarily reflect a shared developmental origin.
Ultimately, although shared Mk and E potential can only be demonstrated through carefully designed single-cell assays, there are transcriptional, immunophenotypic, and functional data in vitro and in vivo for linkage between Mk and E differentiation pathways and the existence of a bipotent MEP among downstream progenitors, although additional distinct routes for Mk development may coexist, as discussed earlier.
Are Mk-E developmental pathways shared between mouse and human?
Studies in mouse models may not always reflect human physiology, and even studies within animal models and tissue types can be hard to compare because of inconsistencies in methods used for cell selection, including antibody panels and gating strategies. This is a particularly important consideration for studying Mks, as mature, terminally differentiated Mks are rare, fragile cells that are difficult to isolate from human bone marrow aspirates, and the majority of studies of human Mks are performed on either ex vivo isolated Mk progenitors or in vitro differentiated cells. The majority of reports of platelet/Mk-biased HSPCs have been conducted in mouse models, although certain aspects of these findings have been recapitulated in studies of human hematopoiesis. Recently, myeloid, E, and Mk differentiation over human development was examined by comparing fetal liver, neonatal cord blood, and adult bone marrow.19 Although multipotent progenitors were found in both the CD38+ and CD38− compartment in fetal liver, the majority of both CD38− and CD38+ cells from adult bone marrow gave unilineage output, indicating that lineage specification may be an earlier event in adult vs fetal human hematopoiesis.19 In single-cell liquid cultures performed in a multilineage cytokine cocktail with MS-5 stromal support cells, the majority of Mks arose in Mk-E or mixed My-Mk-E colonies at all 3 ontological stages, and very few Mk-only colonies were observed particularly from adult bone marrow, where no Mk-only colonies were detected at all using their approach.19 However, this system was not optimized to assess small Mk clones that would be easily missed by FACS analysis.
Similarly, we and others showed that classically defined human Lin−CD34+CD38+CD45RA− (MEP and CMP) cells contain primarily E-only progenitors with smaller fractions of Mk and bipotent cells.54,55 Integrating data from single-cell gene expression profiling and in vitro differentiation assays with index sorting, we demonstrated that individual MEP cells are primarily E biased and give rise to single-lineage output. Classically defined MEP contains 2 major subfractions: cells enriched for Mk-E differentiation but retaining capacity for myeloid differentiation (CD71−CD41− MEP), and E-primed (CD71+CD41− MEP) cells, as well as a rare population of Mk-primed (CD71+CD41+) cells.54 Mk-primed MEPs retained capacity to give rise to E cells in vitro until they acquired CD42, which indicated full commitment to Mk differentiation.54 Lineage switching has also been demonstrated for early blast forming unit-erythroid colonies transferred to secondary cultures stimulated only with thrombopoietin, suggesting that cells destined to give rise to single-lineage progeny retain capacity for alternative lineage differentiation, depending on physiological requirements, up to a certain point of maturation.56 CD41+ cells, largely coexpressing CD42, also exist within the CD123lo/+ CMP compartment in human bone marrow and cord blood progenitors, and are lineage-committed Mk precursor cells.55 Other groups have also reported that the CD41− fraction of immunophenotypic MEP is enriched for bipotent MEP.22 These studies support the presence of a bipotent human MEP (or Pre-MegE), as described in murine HSPCs, although Mk-only colonies occur at significantly lower frequency in human studies compared with mouse. Because of limitations in the assays employed, the exact frequency and absence of myeloid potential in these bipotent Mk-E cells cannot be definitively established.
Moreover, although there is robust evidence of Mk priming of HSCs in the murine system, this remains to be as convincingly demonstrated in normal human hematopoiesis. Of note, expression of Mk molecular markers is substantially higher in murine HSCs than humans. For example, 60% and 10% of LT-HSCs in mice express VWF and CD41, respectively,29,33 in contrast to less than 2% of normal human Lin−CD34+CD38− HSC/MPP, although this may be because of less well-developed purification strategies for human HSCPs or lack of detection, rather than lack of expression, resulting from limited cell numbers studied.57 Similarly, almost 40% of mouse LSK cells express platelet factor 4, which is undetectable in human HSCs.58 Further work is required to detail more precisely the developmental trajectories of megakaryopoiesis in man.
Although mouse and human tissues are most frequently studied, other experimental systems such as zebrafish59 and induced pluripotent stem cell systems60 may also provide important insights and can be used to validate molecular mechanisms suggested in primary tissues. Further, in vivo assays of human HSCs are typically limited to xenotransplants, but further insights can be obtained from lineage tracing in patients who received gene therapy viruses, as was recently reported.61 Although Mk-E differentiation was not specifically examined, this study confirmed that HSCs fulfill distinct roles in steady-state and stress hematopoiesis (eg, the initial period after transplant).61
Although the systems share much homology, caution should be employed in generalizing from mouse to human. There appears to be a stronger Mk bias in murine HSC, although there is insufficient evidence that the Mk-E trajectories are distinct between the species, and any observed differences may simply reflect the assays used.
Do the results of single-cell analyses allow us to redesign a more useful model of mega-erythropoiesis?
A number of different models for the cellular architecture of hematopoiesis have now been proposed. How useful are these models, and what are the implications for understanding normal and malignant hematopoiesis? The robust patterns of lineage reconstitution after single-cell transplants and in lineage tracing studies of unperturbed hematopoiesis undoubtedly confirm that there is an organized, stable, and hierarchical structure to hematopoiesis, including at least correlated trajectories of Mk-E differentiation, rather than continuous emergence of individual lineages in the absence of oligopotent intermediaries.19,20 Substantial heterogeneity exists within the classically defined HSPC populations (HSC, MPP, CMP, MEP), and they are unlikely to represent discrete stages; however, these definitions are useful to immunophenotypically enrich for cells at different stages of differentiation pathways. It may be more valuable to move away from the concept of discrete progenitor stages, and instead conceptualize hematopoiesis as a continuum of differentiation operating in a hierarchical fashion and passing through certain cellular nodes and branch points (Figure 3).
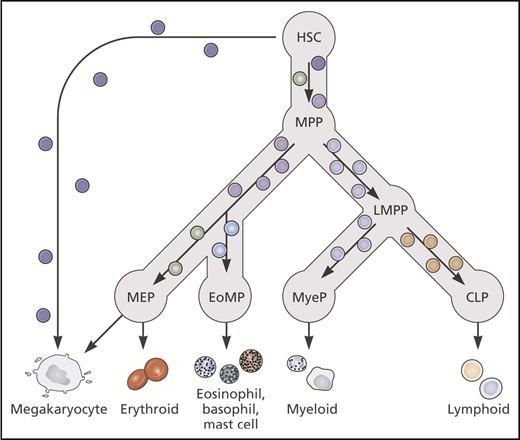
Current literature supports a hierarchical model of hematopoiesis with gradual transition between cellular nodes reflecting increasing priming toward particular lineages and gradual loss of multipotency. Lineage-primed and unipotent cells may exist within multipotent HSC/MPP populations. CLP, common lymphoid progenitor; EoMP, eosinophil-basophil-mast cell progenitor; LMPP, lymphoid-primed multipotent progenitor; MyeP, myeloid progenitor. Professional illustration by Patrick Lane, ScEYEnce Studios.
Current literature supports a hierarchical model of hematopoiesis with gradual transition between cellular nodes reflecting increasing priming toward particular lineages and gradual loss of multipotency. Lineage-primed and unipotent cells may exist within multipotent HSC/MPP populations. CLP, common lymphoid progenitor; EoMP, eosinophil-basophil-mast cell progenitor; LMPP, lymphoid-primed multipotent progenitor; MyeP, myeloid progenitor. Professional illustration by Patrick Lane, ScEYEnce Studios.
In conceptualizing and understanding the utility of the hierarchical model, analogies can be made with sociology, in which different organizational structures are used to explain relationships between and among individuals and groups within a society. Here, a hierarchy is a system in which people or things are arranged according to their importance, and 1 or a few individuals at the top carry all decision-making power. In a nonhierarchical structure, such as a holacracy, leadership is decentralized and decision-making authority is spread across a flat structure. Although there is no question that certain aspects of hematopoiesis are hierarchically organized, the existence of lineage-biased HSC subsets would also incorporate certain aspects of a holacracy model for the organization of single cells within the HSC compartment. In many aspects, this is a more robust model for adult stem cell physiology. In contrast to a rigid hierarchy, in which there is a risk of the leaders abusing power or, for hematopoiesis, mutations arising in primitive HSCs affecting the entire structure (all lineages) below, a holacracy in which HSPCs were configured into self-organizing and self-regulating teams responsible for the production of cells of certain lineages, particularly Mk-E cells, would provide a system able to more rapidly and selectively respond to changing physiological requirements while buffering against malignant outgrowth. Uncoupling of the Mk-E lineages that constitute the main cellular output of hematopoiesis from other myeloid and lymphoid lineages could, in this context, have certain advantages. Given the mounting evidence for lineage-primed HSCs, conceptualizing hematopoiesis as a holacracy, a grouping of discrete but related hierarchies with different lineage biases, may more accurately reflect the true physiological model.
Pathological erythro-megakaryopoiesis and alterations during ageing
Understanding the specific branching points of hematopoiesis is critical to understanding physiological responses to stress and leukemia initiation. In response to inflammation in mice, all populations capable of generating Mks robustly increased their expression of Mk molecular markers, including surface markers (CD41, CD61) and α-granule proteins (VWF, platelet factor 4), and the highest fold change occurred in LT-HSCs, suggesting that a direct path of Mk differentiation from HSC was activated in response to stress.62 This was mediated posttranscriptionally by increased Mk transcript occupancy at ribosomes and enhanced translation of Mk-associated gene transcripts, rather than by altered transcription.62 Lineage priming and the existence of lineage-committed cells within a quiescent stem cell compartment may thereby provide a mechanism for rapid increases in platelet production in response to acute inflammation or acute thrombocytopenia.42
Direct differentiation pathways from HSC to Mk may become more important with aging. Myeloid-biased HSCs expand during aging, and recent evidence suggests that the age-expanded population is specifically platelet- rather than pan-myeloid-biased. CD41 expression on LSK cells increases 10-fold from young (2 months) to old (16 months) mice.63 Both the number of platelet-specific genes and their expression level increases in aged HSCs, together with a preferential expansion of VWF+ platelet-primed HSCs, by almost 50-fold, as well as an increase in the proportion of these cells that have platelet-biased clonogenic output.64 The increasing myeloid and Mk bias of HSCs has implications for understanding clonal hematopoiesis and myeloid malignancies. For example, self-renewing and lineage-biased yet multipotent HSC subsets could give rise to clonal hematopoiesis.65 Further, increased Mk-priming of aged HSCs may underlie the pathologically increased megakaryopoiesis that occurs in myeloproliferative neoplasms that increase in incidence with age.57 Understanding the specific target cell for oncogenic mutations may explain key features of disease pathophysiology and heterogeneity, and reveal novel targets for therapy.66,67
Future directions
Further insights into the molecular regulation of the cellular pathways of Mk-E differentiation may reveal novel targets for therapy of disorders characterized by anemia and/or thrombocytopenia, as well as for improving recovery after bone marrow transplant and for new methods to generate red cells and platelets for transfusion or after genome editing. Specific HSPC subfractions could be purified and targeted for genome editing to replenish blood cells of Mk or E lineage.
In addition to single-cell transcriptomics and functional studies, a multilayered single-cell-omics approach may be necessary to determine not only the intrinsic properties of individual cells but also their spatial context in situ (eg, Mk-biased HSC may occupy specific hematopoietic niches) and combined multiomic approaches to simultaneously examine DNA, RNA, and epigenetics within the same single cell.68 These studies may further unmask particular facets of lineage bias. Transcriptome analyses typically only provide snapshots of cellular activity, although dynamic information can be inferred by computational approaches.69 A novel approach was recently published in which stably expressed genes and recently activated or repressed genes are distinguished by the ratio of immature or unspliced to spliced mRNA, thereby inferring the RNA velocity of each gene to suggest the transcriptional changes that are occurring within each cell.70
To conclude, the current literature supports that in normal hematopoiesis, the majority of Mks arise via a shared trajectory with the E lineage. The sharing is the codependence on common transcription factors (eg, GATA1) and growth factors-receptor pairs (eg, SCF-cKit). HSCs can be Mk-primed, but retain multipotency and capacity to change path if required. In addition, distinct pathways for direct differentiation of Mk from HSC may coexist and could be important in situations of increased physiological requirements, or emergency hematopoiesis, or in disease states. Further work at single-cell resolution using multiomic approaches, lineage tracing, and examining Mk-E biased subsets within their physiological context will undoubtedly improve both our understanding of normal hematopoiesis and our ability to manipulate this in pathology.
Authorship
Contribution: B.P. and A.J.M. reviewed the relevant literature, wrote the manuscript, and drew the figures.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Bethan Psaila, Molecular Haematology Unit, University of Oxford, Medical Research Council (MRC) Weatherall Institute of Molecular Medicine, John Radcliffe Hospital, Headington, Oxford OX3 9DS, United Kingdom; e-mail: bethan.psaila@ndcls.ox.ac.uk; and Adam J. Mead, Molecular Haematology Unit, University of Oxford, MRC Weatherall Institute of Molecular Medicine, John Radcliffe Hospital, Headington, Oxford OX3 9DS, United Kingdom; e-mail: adam.mead@imm.ox.ac.uk.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal