

Key Points
Kiaa0157 deficiency expands the HSC pool and increases reconstitution potential.
BRISC deficiency increases JAK2 K63-ubiquitination, enhances its association with TPO receptor at the cell membrane, and promotes signaling.

Abstract
Hematopoietic stem cell (HSC) homeostasis is controlled by cytokine receptor–mediated Janus kinase 2 (JAK2) signaling. We previously found that JAK2 is promptly ubiquitinated upon cytokine stimulation. Whether a competing JAK2 deubiquitination activity exists is unknown. LNK is an essential adaptor protein that constrains HSC expansion through dampening thrombopoietin (TPO)–induced JAK2 signaling. We show here that a LNK-associated lysine-63 (K63)–deubiquitinating enzyme complex, Brcc36 isopeptidase complex (BRISC), attenuates HSC expansion through control of JAK2 signaling. We pinpoint a direct interaction between the LNK SH2 domain and a phosphorylated tyrosine residue in KIAA0157 (Abraxas2), a unique and defining BRISC component. Kiaa0157 deficiency in mice led to an expansion of phenotypic and functional HSCs. Endogenous JAK2 and phospho-JAK2 were rapidly K63-ubiquitinated upon TPO stimulation, and this action was augmented in cells depleted of the BRISC core components KIAA0157, MERIT40, or BRCC36. This increase in JAK2 ubiquitination after BRISC knockdown was associated with increased TPO-mediated JAK2 activation and protein levels, and increased MPL receptor presence at the cell surface. In addition, BRISC depletion promoted membrane proximal association between the MPL receptor and pJAK2/JAK2, thus enhancing activated JAK2/MPL at the cell membrane. These findings define a novel pathway by which K63-ubiquitination promotes JAK2 stability and activation in a proteasome-independent manner. Moreover, mutations in BRCC36 are found in clonal hematopoiesis in humans. This research may shed light on the mechanistic understanding of a potential role of BRCC36 in human HSCs.
Introduction
Hematopoietic stem cell (HSC) homeostasis is controlled by cytokine-mediated signaling. One such signaling axis is thrombopoietin (TPO) and its receptor Mpl that maintains HSC homeostasis and promotes self-renewal.1,2 TPO binding to Mpl activates Janus kinase 2 (JAK2) at the plasma membrane, triggering multiple signaling events.3,4 JAK2-deficient hematopoietic cells fail to respond to an array of hematopoietic cytokines, revealing its essential role in cytokine receptor signaling and hematopoietic development. We and others have previously reported that Lnk (also called SH2B3) is an essential adaptor protein that constrains HSC expansion through dampening TPO/Mpl-induced JAK2 signaling.5-7 Lnk−/− mice have a markedly expanded HSC pool, with superior reconstitution ability and increased self-renewal.5,8 In search of mechanisms by which LNK regulates JAK2 signaling in HSCs, we identified a LNK-associated deubiquitinating enzyme (DUB) complex.9
Polyubiquitination (polyUb) is a posttranslational modification involving the covalent conjugation of Ub chains to specific protein substrates. Ub has 7 lysines (K6, K11, K27, K29, K33, K48, and K63), all of which can participate in peptide bond formation.10,11 K48Ub is the canonical form that targets proteins for degradation through the proteasome.11 In contrast, K63Ub does not target proteins to the proteasome but rather mediates various biological processes, including DNA repair,12,13 protein trafficking,14 autophagy,15 and signal transduction.16 An in vivo role of K63Ub in hematopoiesis has been previously suggested based on the observation that the loss of Ubc13 (the Ub-conjugating enzyme specific for K63Ub chains) in mice leads to hematopoietic failure owing to loss of HSCs and progenitors (HSPCs).17 However, how K63Ub affects hematopoiesis or HSC function has not been well established.
Ubiquitination is a reversible process, as DUBs rapidly de-conjugate ubiquitinated substrates.18 Brcc36, a K63Ub-specific DUB, belongs to a small class of Zn2+-dependent isopeptidases commonly found in association with large protein complexes.19,20 Indeed, Brcc36 is a component of 2 complexes, 1 nuclear and 1 cytoplasmic, that share common components but are distinct in their localization and function.13,21-23 The cytoplasmic Brcc36 isopeptidase complex (BRISC) complex consists of 4 subunits: 1 unique component KIAA0157 (also called Abraxas2 or Abro1) and 3 shared components (Brcc45/MERIT40/Brcc36) that are also in the nuclear Abraxas1 complex.23
We previously showed that MERIT40 (or BABAM1), the scaffold subunit of both complexes, attenuates HSC expansion as well as TPO/Mpl-mediated JAK2 signaling.9 Merit40 (M40) deficiency increases the size of phenotypic and functional HSC pools. M40 deficiency triggers hypersensitivity to TPO stimulation, and the stem cell phenotypes are abrogated on a background null for the TPO receptor Mpl. These results establish MERIT40-containing DUB complexes as novel regulators of HSC expansion. However, how MERIT40 regulates cytokine-JAK signaling is unknown. Moreover, how K63Ub regulates cytokine-JAK2 signaling is poorly established.
Here we show that Lnk binds to the unique BRISC subunit KIAA0157 through a phosphorylation-dependent interaction to control K63Ub-regulated JAK2 signaling in HSCs. This study shows that BRISC K63Ub-specific deubiquitinating activity on JAK2 is critical for its cellular activation and HSC homeostasis, implicating nondegradative deubiquitination in JAK2-driven stem cell function.
Materials and methods
Mice and cell lines
C57/BL6 (CD45.2), SJL (CD45.1), and F1 (CD45.1/CD45.2) breeding pairs were from Jackson Laboratories and maintained at the in-house facility. Kiaa−/− mice on the C57/B6 background were generated previously.21 This study was conducted under an approved protocol from the Institutional Animal Care and Use Committee of the Children’s Hospital of Philadelphia. All cell lines were originally purchased from ATCC. HEL cells were maintained in RPMI1640 containing 10% heat-inactivated calf serum (Thermo Fisher Scientific); TF-1 cell media were supplemented with 2 ng/mL human granulocyte-macrophage colony-stimulating factor. 32D and BaF3 cells were maintained in RPMI containing 10% calf serum and 10% WEHI-3B cell supernatant as a source of interleukin-3. HEK-293T cells were maintained in DMEM containing 10% calf serum.
Constructs
Retroviral MIG-Lnk (MSCV-Lnk-ires-GFP), pOZ-Lnk (pMX-Flag/HA-Lnk-ires-IL-2R), and the pOZ-Lnk 2SA mutant constructs were generated as previously described.5,24 The pOZ-hLNK mutants were generated through site-directed mutagenesis using a QuikChange Kit and confirmed by sequencing (Stratagene). pOZ-BRCC36 and KIAA0157 were generated previously.25 Lentiviral pLKO.1-puro short hairpin RNA (shRNA) to Luc or M40 were purchased from Open Biosystems. miR30-based shRNA against KIAA0157 (CAGAGCCTTCTAATAGTGAAT) and BRCC36 (GCACAGAGAAGGAGGAAGTAA) were constructed into PIG (MSCV-puro-ires-GFP) retroviral vector.26
Transfection, viral packaging, and infection
293T cells were transfected with plasmids using FuGene6 reagents; 2 days later, cells were lysed for biochemical studies. Retroviral or lentiviral constructs were cotransfected with pCL-Ecotropic or pCL-Amphotropic helper viral packaging plasmids into 293T cells, and the resulting supernatants were collected 2 days later. All studies using BaF3, HEL, or TF-1 cells were stable cell lines generated by spin-infection with the desired viral supernatant containing 10 μg/mL polybrene (MilliporeSigma) at 640g for 1 hour at 30°C. Cells were either subsequently sorted for green fluorescent protein fluorescent or surface markers (hIL2R for pOZ vector) or selected for drug resistance (puromycin 5 μg/mL).
Immunoprecipitation, Western blot analysis, and protein half-life assays
TF-1/MPL or BaF3/MPL cells were starved for 1 to 3 hours in RPMI 0.5% bovine serum albumin and stimulated with 100 ng/mL recombinant human TPO (PeproTech #300-18) for the indicated times, or stimulated with a graded concentration of TPO for the indicated times. For cycloheximide chase assay, TF-1/MPL cells were starved, then treated with 40 μg/mL cycloheximide in the absence or presence of 100 ng/mL human TPO for the indicated time points. Protein half-lives were evaluated by using western blot (WB) analysis.
For WB analysis, cells were directly lysed in lithium dodecyl sulfate reagent (Thermo Fisher Scientific), lysates resolved on Tris-Glycine polyacrylamide gels (National Diagnostics), and transferred to nitrocellulose membranes (Protran). The WB analyses were conducted with the following antibodies: anti-JAK2 (Cell Signaling #3230), phospho-JAK2 (Cell Signaling #3776), phosphotyrosine (MilliporeSigma #05-321), Ubiquitin (Cell Signaling #3936), K63Ub monoclonal (eBioscience), BRCC36 (Bethyl Laboratories #A302), KIAA0157/ABRO1 (Bethyl Laboratories #A301), BRCC45,25 MERIT40,12 Lnk,9 Myc (Cell Signaling, clone 9B11), phospho-STAT5 (Cell Signaling #9351), STAT5 (Santa Cruz Biotechnology #sc-835), or ACTIN (Santa Cruz Biotechnology #sc-1616).
For immunoprecipitation (IP), TF-1 cells were lysed in IP buffer (10 mM Tris, pH 7.4, 150 mM NaCl, 0.5% NP-40, 1 mM NaF, 1 mM Na3VO4, phenylmethylsulfonyl fluoride, and protease inhibitor cocktail) at 4°C for 30 minutes. After clarification by centrifugation, cell lysates were precleared with protein A/G beads for 1 hour, then incubated with anti-Lnk,9 Flag (MilliporeSigma #F2426), Myc (9E10), or JAK2 (MilliporeSigma #06-255) antibodies.
Mass spectrometry
32D-BCR/ABL (32D-B/A) cells that stably expressed Flag/HA-tagged Lnk (pOZ-FH-Lnk or 2SA mutant) were established as previously described.24 Cytoplasmic fractions were isolated and precipitated first with anti-Flag resin (M2, MilliporeSigma #F2426), eluted with 3X Flag peptides, followed by a second IP with anti-HA (1:1 ratio mix of HA-7 and HA-11 antibodies) agarose. A small aliquot of precipitated lysates was resolved on NuPAGE gels (Thermo Fisher Scientific) and the protein bands visualized with silver staining. The rest of the lysates were then resolved and stained with Colloidal Blue (Thermo Fisher Scientific), and gel slices were excised and submitted to the Harvard Taplin Mass Spectrometry Facility for protein identification.
Ubiquitin pulldown assay
Ubiquitin pulldown assays were performed according to the manufacturer’s protocol. Briefly, cells were lysed in the lysis buffer (100 mM Tris-HCL, pH 8.0, 150 mM NaCl, 5mM EDTA, 1% NP-40, 0.5% Triton-X 100, protease inhibitor cocktail [Roche], 100 μM PR-619 [LifeSensors], 4 mM 1,10-phenanthroline [o-PA] [Mallinckrodt Chemicals], 4 mM N-Ethylmaleimide [MilliporeSigma], 100 nM Flag K63-TUBE [UM604], LifeSensors) for 1 hour. Cell lysates were clarified by centrifugation and the supernatant diluted fourfold, then incubated at 4°C for 2 hours to allow for binding of Flag-K63-TUBE to polyUb chains. Flag M2 affinity resins were then added to cell lysates and incubated for 2 hours at 4°C. Precipitates were eluted with lithium dodecyl sulfate loading buffer and then subjected to WB analysis. For pulldown of total ubiquitinated proteins, cells were pretreated with MG132 for 100 minutes in starvation media, then stimulated with or without TPO for 20 minutes.
Cell proliferation assay
To measure TF-1 cell proliferation, triplicate samples of cells were seeded at 2 × 105/mL in 96-well plates and cultured with different concentrations of cytokines. Three days later, 3-(4, 5-dimethylthiazolyl-2)-2, 5-diphenyltetrazolium bromide (0.5 mg/mL; MilliporeSigma) was added to the cultures for 3 hours, and live cell numbers were measured according to the manufacturer’s instructions.
Mpl internalization
TF1-hMpl were starved, then stimulated with 100 ng/mL human TPO for 0, 10, 20, 30, and 60 minutes. Cells were immediately transferred to ice and fixed in 1% paraformaldehyde (EMS Biosciences) for 30 minutes. Cells were then incubated with Fc-blocker (BD Biosciences) for 15 minutes, followed by staining with anti-human CD110 (Mpl)-phycoerythrin (PE) antibody (clone 1.6.1; BD Biosciences) for 45 minutes. Subsequently, stained cells were subjected to flow cytometry (FACSCanto) and for the analysis of the PE geometric mean fluorescent intensity on the FlowJo software.
Cell surface IP
Cells were then placed on ice-cold blocking buffer (PBS + 0.5% bovine serum albumin) for 30 minutes, followed by incubation with 5 μl/mL anti-CD110 antibodies (clone 1.6.1; BD Biosciences) for 1 hour at 4°C. After extensive washes with blocking buffer to remove excess antibodies that bind to the cell surface MPL, cells were lysed in 1% NP-40 followed by incubation with protein G agarose for 90 minutes at 4°C.
Flow cytometric analysis of HSPC subsets
HSPC analysis was performed as previously described.5,24,27 Briefly, bone marrow (BM) cells were stained with FITC-CD45.1, APC-CD45.2, biotinylated lineage cocktail, PerCP-Cy5.5-Sca1, APC-Cy7-Kit, PE-Cy7-CD150, FITC-CD48, APC-CD34, and PE-Flk2 antibodies (eBioscience), followed by streptavidin-PE-Texas Red secondary antibodies (Thermo Fisher Scientific). Cells were resuspended in 4′,6-diamidino-2-phenylindole–containing fluorescence-activated cell sorter buffer and subjected to flow cytometric analysis on an LSRFortessa (BD Biosciences). Data were analyzed by using FlowJo software.
Limiting dilution BM transplantation
Serially diluted total BM cells (CD45.2) from wild-type (WT) or Kiaa−/− mice were mixed with 3 × 105 competitor BM cells (CD45.1) and injected retro-orbitally into lethally irradiated (a split dose of 10 Gy orthovoltage X-ray, Precision X-ray Inc.) F1 recipient mice. Sixteen weeks after injection, percent donor-derived cells in peripheral blood was determined by flow cytometry.5 Briefly, cells were stained with antibodies to different lineages: PE-Gr1 and APC-Mac1 for myeloid cells, PE-CD4 and CD8 for T cells, and APC-B220 for B cells, along with FITC-CD45.1 and APC-Cy7-CD45.2 to differentiate donor cells from competitor/host cells.
Statistical analysis
Two-tailed Student t tests were used. P < .05 was considered significant.
Results
LNK interacts with BRISC complex through KIAA0157
We recently reported that LNK binds to MERIT40, a core protein within the BRISC complex.9 Because LNK also robustly binds the ubiquitous 14-3-3 family proteins,24 we first set out to eliminate the possibility of an indirect interaction between BRISC and LNK through the abundant 14-3-3 proteins present in the cell. We generated 32D-B/A cells5,24 expressing with FLAG-HA-tagged WT Lnk or the S13A/S129A (2SA) mutant that is deficient in 14-3-3 interaction.24 We then performed tandem affinity purification of double epitope-tagged Lnk followed by mass spectrometric analysis. All 4 members of the BRISC complex (KIAA0157, BRCC36, BRCC45, and MERIT40) were identified in both WT and 2SA Lnk expressing cells, whereas the Lnk2SA mutant disrupted the Lnk/14-3-3 interaction, suggesting that Lnk binds to BRISC independently of 14-3-3 (Figure 1A; supplemental Table 1, available on the Blood Web site).
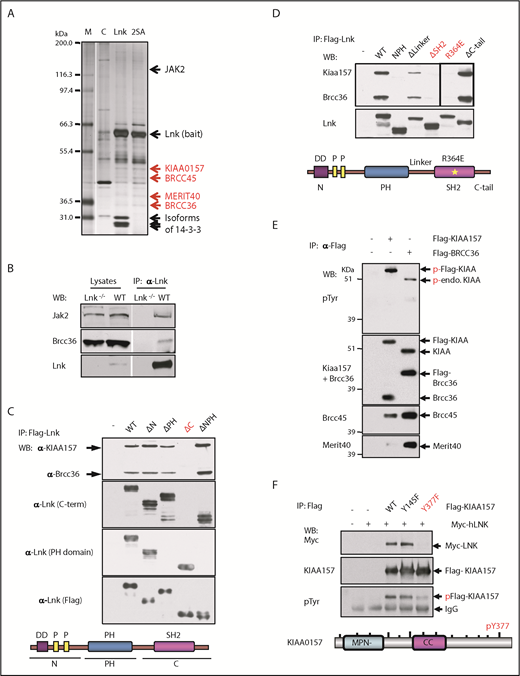
The BRISC complex is a novel Lnk binding partner. (A) Identification of a novel Lnk–BRISC interaction. Cytoplasmic protein extracts of 32D-B/A parental cells (C) or 32D cells expressing Flag/HA-tagged WT Lnk or Lnk2SA mutant (2SA) deficient in 14-3-3 interaction, were immunoprecipitated with anti-FLAG and anti-HA antibodies sequentially. A small aliquot of precipitates was resolved in sodium dodecyl sulfate–polyacrylamide gel electrophoresis and protein bands visualized by silver stain as shown. Subsequently, a large aliquot of precipitates was stained by using Coomassie stains and protein bands identified by using mass spectrometry. Subunits of the BRISC complex are indicated in red. (B) Confirmation of Lnk–BRISC interaction. Lysates from WT and Lnk−/− spleens were either directly subjected to WB analysis (left) or precipitated with anti-Lnk antibodies followed by WB analysis (right) with indicated antibodies. (C) The Lnk C terminus binds to BRISC. Lysates from 32D-B/A cells stably expressing Flag-WT or mutant Lnk were precipitated with anti-Flag antibodies followed by WB analysis. The bottom illustrates the Lnk structure. N terminus (N) contains a dimerization domain and 2 proline-rich regions. (D) The Lnk SH2 domain binds to BRISC. 32D-B/A cells stably expressing Flag-WT Lnk or constructs containing mutations or small deletions in the C terminus of Lnk were generated. IP-WB analyses were performed as in panel C. The bottom illustrates the Lnk structure and indicates the mutated regions. *R364E point mutation. (E) KIAA0157 is tyrosine phosphorylated in hematopoietic cells. HEL cells expressing either Flag-tagged KIAA0157 or BRCC36 were precipitated with anti-Flag antibodies followed by WB analysis with antibodies to phospho-tyrosine (pTyr) and the BRISC components as indicated. (F) Lnk binds to pY377 in KIAA0157. HEL cells expressing Flag-KIAA0157 or mutants, along with Myc-Lnk, were lysed and precipitated with anti-Flag antibodies followed by WB analysis with the indicated antibodies. 4G10 antibodies recognize pTyr. The bottom illustrates the KIAA0157 structure. Δ, deletion; CC, coil-coiled domain that interacts with BRCC36; endo, endogenous; F, phenylalanine; FH, Flag/HA-tagged; M, marker; P, phosphorylated form; Y, tyrosine residues.
The BRISC complex is a novel Lnk binding partner. (A) Identification of a novel Lnk–BRISC interaction. Cytoplasmic protein extracts of 32D-B/A parental cells (C) or 32D cells expressing Flag/HA-tagged WT Lnk or Lnk2SA mutant (2SA) deficient in 14-3-3 interaction, were immunoprecipitated with anti-FLAG and anti-HA antibodies sequentially. A small aliquot of precipitates was resolved in sodium dodecyl sulfate–polyacrylamide gel electrophoresis and protein bands visualized by silver stain as shown. Subsequently, a large aliquot of precipitates was stained by using Coomassie stains and protein bands identified by using mass spectrometry. Subunits of the BRISC complex are indicated in red. (B) Confirmation of Lnk–BRISC interaction. Lysates from WT and Lnk−/− spleens were either directly subjected to WB analysis (left) or precipitated with anti-Lnk antibodies followed by WB analysis (right) with indicated antibodies. (C) The Lnk C terminus binds to BRISC. Lysates from 32D-B/A cells stably expressing Flag-WT or mutant Lnk were precipitated with anti-Flag antibodies followed by WB analysis. The bottom illustrates the Lnk structure. N terminus (N) contains a dimerization domain and 2 proline-rich regions. (D) The Lnk SH2 domain binds to BRISC. 32D-B/A cells stably expressing Flag-WT Lnk or constructs containing mutations or small deletions in the C terminus of Lnk were generated. IP-WB analyses were performed as in panel C. The bottom illustrates the Lnk structure and indicates the mutated regions. *R364E point mutation. (E) KIAA0157 is tyrosine phosphorylated in hematopoietic cells. HEL cells expressing either Flag-tagged KIAA0157 or BRCC36 were precipitated with anti-Flag antibodies followed by WB analysis with antibodies to phospho-tyrosine (pTyr) and the BRISC components as indicated. (F) Lnk binds to pY377 in KIAA0157. HEL cells expressing Flag-KIAA0157 or mutants, along with Myc-Lnk, were lysed and precipitated with anti-Flag antibodies followed by WB analysis with the indicated antibodies. 4G10 antibodies recognize pTyr. The bottom illustrates the KIAA0157 structure. Δ, deletion; CC, coil-coiled domain that interacts with BRCC36; endo, endogenous; F, phenylalanine; FH, Flag/HA-tagged; M, marker; P, phosphorylated form; Y, tyrosine residues.
To further verify this interaction, we isolated spleen lysates from WT or Lnk−/− mice. Endogenous Lnk indeed co-precipitated with BRCC36, the K63 DUB component of the BRISC complex (Figure 1B). To determine the necessary domains in Lnk required for its interaction with BRISC, we established 32D-B/A cells stably expressing either Flag-WT Lnk or various deletion mutants. Neither the N terminus nor the PH domain of the LNK protein was required for BRISC interaction; however, the C terminus was critical for LNK–BRISC interaction (Figure 1C). This scenario was also confirmed by using human LNK complementary DNA and corresponding mutants stably expressed in a human erythroleukemia cell line, HEL cells (supplemental Figure 1A). We then mutated the SH2 domain near the C terminus, the C terminus tail, or the linker region between the PH and SH2 domains (Figure 1D). The Lnk mutant lacking its SH2 domain (ΔSH2) failed to coprecipitate (IP) with endogenous BRISC members. Furthermore, Lnk R364E, an SH2 domain point mutant that abolishes Lnk-mediated growth inhibition,28 did not co-precipitate with BRISC, indicating that Lnk interacts with BRISC through its SH2 domain.
Because SH2 domains specifically interact with phospho-tyrosine residues, we first investigated which BRISC member(s) are phosphorylated. Lysates from HEL cells stably expressing either Flag-KIAA0157 or Flag-BRCC36 were precipitated with anti-Flag resin. Flag IP pulled down all 4 BRISC components (Figure 1E). We then subjected the IPs to WB analysis with anti–phospho-tyrosine antibodies. Both endogenous and exogenous KIAA0157 were tyrosine phosphorylated but other BRISC components were not. We next sought to pinpoint the specific tyrosine residues in KIAA0157 that interact with Lnk by interrogating the consensus motif (pY(V/I/E)xL) we previously identified.29 Tyrosine to phenylalanine mutants in 2 candidate Ys (Y377F and Y145F) were generated and stably coexpressed with Myc-Lnk in 32D cells. Y377F but not Y145F mutation in KIAA0157 markedly reduced tyrosine phosphorylation of KIAA (Figure 1F). Co-IP experiments revealed that pY377 residue in KIAA0157 is required for BRISC interaction with Lnk in both HEL and murine 32D cells (supplemental Figure 1B). The SH2B family members, including LNK (SH2B3), reportedly formed dimers or oligomers.30 Our data indicate that LNK bridges the interaction between BRISC and JAK2.
KIAA157 deficiency in mice expands long-term HSCs
Our data suggest that LNK specifically interacts with the BRISC complex via KIAA0157 (Abraxas2). The DUB component of BRISC (BRCC36) is also associated with a closely related nuclear complex containing KIAA0157 paralog Abraxas1. The nuclear complex, also called BRCA1-A complex, interacts with RAP80 and brings BRCA1 to the DNA damage site.13,22,31,32 Kiaa0157-deficient cells exhibited an unaffected DNA damage response as evidenced by normal recruitment of BRCA1 and RAP80 to the DNA damage site (supplemental Figure 2). This finding is in contrast to M40-deficient cells, which are critical for the integrity of both BRISC and BRCA1-A complexes.12 More importantly, Kiaa0157−/− mice are not sensitive to MMC, whereas Merit40−/− mice exhibit sensitivity to MMC.33 These data confirm that KIAA deficiency specifically abrogates BRISC while leaving the nuclear BRCA1-A complex intact.
We previously showed that Merit40−/− mice harbor an expanded HSC pool with increased repopulating ability.9 Our goal was to investigate if this HSC function is accounted for by BRISC using Kiaa0157−/− (Kiaa−/−) mice, as KIAA0157 is the unique BRISC component. Deletion of Kiaa0157 markedly reduced BRISC components BRCC36 and MERIT40 in the BM (Figure 2A), indicating BRISC is the predominant complex in the BM.
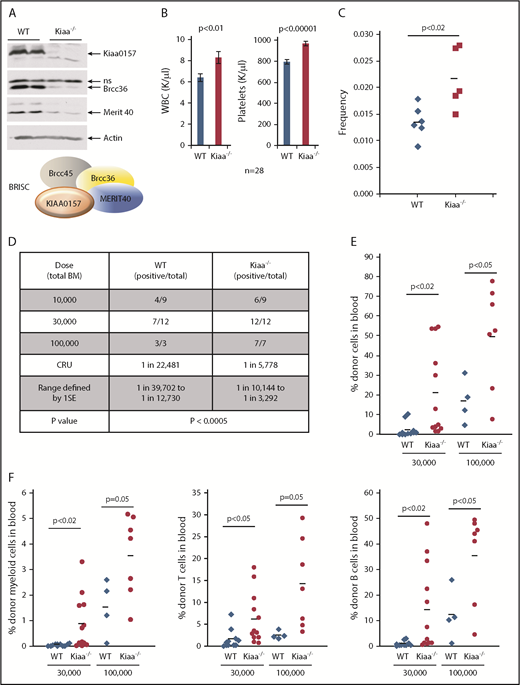
KIAA0157 deficiency leads to an expansion of phenotypic and functional HSCs. (A) KIAA0157 depletion disrupts the BRISC complex. WB analysis of BM extracts from WT and Kiaa0157−/− (Kiaa−/−) mice. (B) White blood cell (WBC) counts and platelet counts in the peripheral blood of WT and Kiaa−/− mice are shown. N = 28. (C) Frequency of long-term HSCs in WT and Kiaa−/− mice as determined by flow cytometry. Long-term HSC is defined as CD34-Flk2-CD150+LSK. Each symbol represents an individual mouse, and the horizontal lines indicate the mean values. P value was determined by 2-tailed Student t test. (D-F) Functional HSC frequency in WT and Kiaa−/− mice as determined by limiting dilution BM transplantation. Percentage of donor-derived cells in peripheral blood 16 weeks after BM transplantation were analyzed by flow cytometry. Data from 2 to 3 independent experiments were combined. (D) The results are presented as number of positively engrafted mice vs total number of mice analyzed for the indicated doses. Positive engraftment was defined as >1% donor-derived cells in the peripheral blood. Reconstitution frequency and statistical analysis were calculated using ELDA software (http://bioinf.wehi.edu.au/software/elda/). (E-F) Donor chimerism in peripheral blood of the recipient mice transplanted with 3 ×104 and 1 × 105 donor cells along with competitors are shown. (E) Total leukocyte reconstitution. (F) Donor reconstitution in different blood lineages. Each symbol represents an individual mouse; horizontal lines indicate the mean value in each group. P values were calculated by using the 2-tailed Student t test. 1SE, one standard deviation; CRU, competitive repopulating unit; ns, nonspecific.
KIAA0157 deficiency leads to an expansion of phenotypic and functional HSCs. (A) KIAA0157 depletion disrupts the BRISC complex. WB analysis of BM extracts from WT and Kiaa0157−/− (Kiaa−/−) mice. (B) White blood cell (WBC) counts and platelet counts in the peripheral blood of WT and Kiaa−/− mice are shown. N = 28. (C) Frequency of long-term HSCs in WT and Kiaa−/− mice as determined by flow cytometry. Long-term HSC is defined as CD34-Flk2-CD150+LSK. Each symbol represents an individual mouse, and the horizontal lines indicate the mean values. P value was determined by 2-tailed Student t test. (D-F) Functional HSC frequency in WT and Kiaa−/− mice as determined by limiting dilution BM transplantation. Percentage of donor-derived cells in peripheral blood 16 weeks after BM transplantation were analyzed by flow cytometry. Data from 2 to 3 independent experiments were combined. (D) The results are presented as number of positively engrafted mice vs total number of mice analyzed for the indicated doses. Positive engraftment was defined as >1% donor-derived cells in the peripheral blood. Reconstitution frequency and statistical analysis were calculated using ELDA software (http://bioinf.wehi.edu.au/software/elda/). (E-F) Donor chimerism in peripheral blood of the recipient mice transplanted with 3 ×104 and 1 × 105 donor cells along with competitors are shown. (E) Total leukocyte reconstitution. (F) Donor reconstitution in different blood lineages. Each symbol represents an individual mouse; horizontal lines indicate the mean value in each group. P values were calculated by using the 2-tailed Student t test. 1SE, one standard deviation; CRU, competitive repopulating unit; ns, nonspecific.
Kiaa−/− mice exhibited elevated white blood cell and platelet counts, similar to Merti40−/− mice (Figure 2B). Importantly, Kiaa−/− mice possessed a higher frequency of long-term HSCs, through stringent phenotypic markers, CD34-Flk2-CD150+LSK (LSK as Lineage-Sca1+c-Kit+) (Figure 2C). To functionally quantify HSC frequency in Kiaa−/− mice, competitive limiting dilution BM transplantation was used. Serially diluted BM cells from either WT or Kiaa−/− mice were transplanted along with competitor BM cells (Figure 2D-F). The results revealed that Kiaa−/− mice not only had an increased frequency of functional HSCs but also displayed more robust recipient repopulation in all blood lineages in the transplanted mice. Hence, our data strongly support that BRISC deficiency leads to an expansion of the phenotypic and functional HSC pool.
BRISC depletion increases JAK2 K63-polyubiquitination
We recently showed that endogenous JAK2 is K63-ubiquitinated upon TPO stimulation.26 Because BRISC specifically cleaves K63-polyubiquitin (K63-polyUb) chains of its substrates, and LNK constrains HSC expansion by regulating the TPO/Mpl/JAK2 signaling,5 we first explored the ubiquitination kinetics of JAK2. We stimulated BaF3/hMPL cells over a 60-minute time course along with pretreatment with proteasome inhibitor MG132. K63-polyUb proteins were pulled down by using FLAG-K63-TUBE that shows an >1000-fold preference over other Ub chains,34 followed by IP with anti-FLAG Sepharose. WB analysis revealed that JAK2 and pJAK2 were K63-polyubiquitinated. This finding correlated with JAK2 phosphorylation, peaking at 10 to 20 minutes after TPO stimulation (Figure 3A, left panel). In parallel, we pulled down JAK2 with anti-JAK2 antibodies, then subjected the blot to Far-WB analysis with TUBE1-biotin (Figure 3A, right panel), which detects both K63 and K48 poly-Ub chains. We observed a TPO-induced JAK2 ubiquitination that was sustained over 60 minutes. Monoclonal antibodies to K63Ub suggested a prompt but transient induction of JAK2 K63-ubiquitination, consistent with the K63TUBE pulldown. To further confirm that JAK2 is K63-ubiquitinated, we co-transfected Myc-JAK2 along with HA-WT Ub or Ub mutants into 293T cells. Co-IP experiments showed that JAK2 ubiquitination is significantly reduced in cells expressing K63-deficient and K48-deficient Ub, whereas K63-only Ub but not K48-only Ub enabled JAK2 polyubiquitination (supplemental Figure 3). These results suggest that JAK2 is largely modified through K63Ub in addition to K48-Ub.35
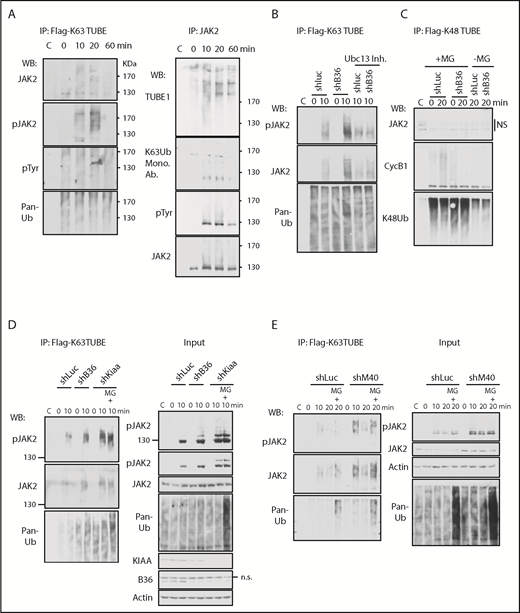
BRISC depletion enhances JAK2 K63-ubiquitination. (A) K63-ubiquitination of JAK2 correlates with its activity. BaF3/hMpl cells were stimulated with TPO for indicated times. Left panel: Lysates were incubated with Flag-K63TUBE followed by IP with anti-Flag beads. The blots were probed with indicated antibodies. Right panel: Lysates were precipitated with anti-JAK2 serum, and the blot was probed with TUBE1-biotin followed by detection with streptavidin-conjugated horseradish peroxidase secondary antibody for Far-Western detection. The blot was then stripped and probed with indicated antibodies. (B-D) BRISC depletion increases JAK2 K63Ub. (B) TF-1/MPL cells expressing shRNA-mediated knockdown to either controls Luciferase (Luc) or Brcc36 (B36) were pretreated with or without Ubc13 inhibitors and stimulated with TPO for 0 or 10 min. Flag-K63TUBE pulldown was performed as in panel A and blotted with indicated antibodies. (C) TF-1/MPL cells expressing either shRNA to Luc or B36 were pretreated with or without MG132 and stimulated with TPO for 0 or 20 min. Cell lysates were pulled down with Flag-K48TUBE followed by WB analysis with indicated antibodies. (D-E) K63Ub-TUBE pulldowns were performed similarly in TF-1/MPL cells expressing either shRNA to Luc, B36, or KIAA (D) or 32D/Mpl cells expressing either shRNA to Luc or M40 (E). WB analysis of total cell lysates is indicated at “Input” in the right panels. C, control immunoglobulin G; Mono. Ab., monoclonal antibody.
BRISC depletion enhances JAK2 K63-ubiquitination. (A) K63-ubiquitination of JAK2 correlates with its activity. BaF3/hMpl cells were stimulated with TPO for indicated times. Left panel: Lysates were incubated with Flag-K63TUBE followed by IP with anti-Flag beads. The blots were probed with indicated antibodies. Right panel: Lysates were precipitated with anti-JAK2 serum, and the blot was probed with TUBE1-biotin followed by detection with streptavidin-conjugated horseradish peroxidase secondary antibody for Far-Western detection. The blot was then stripped and probed with indicated antibodies. (B-D) BRISC depletion increases JAK2 K63Ub. (B) TF-1/MPL cells expressing shRNA-mediated knockdown to either controls Luciferase (Luc) or Brcc36 (B36) were pretreated with or without Ubc13 inhibitors and stimulated with TPO for 0 or 10 min. Flag-K63TUBE pulldown was performed as in panel A and blotted with indicated antibodies. (C) TF-1/MPL cells expressing either shRNA to Luc or B36 were pretreated with or without MG132 and stimulated with TPO for 0 or 20 min. Cell lysates were pulled down with Flag-K48TUBE followed by WB analysis with indicated antibodies. (D-E) K63Ub-TUBE pulldowns were performed similarly in TF-1/MPL cells expressing either shRNA to Luc, B36, or KIAA (D) or 32D/Mpl cells expressing either shRNA to Luc or M40 (E). WB analysis of total cell lysates is indicated at “Input” in the right panels. C, control immunoglobulin G; Mono. Ab., monoclonal antibody.
We next investigated the impact of BRISC in JAK2 K63-polyUb using shRNA-mediated knockdown of various components of BRISC, BRCC36, KIAA0157, and MERIT40 (Figure 3B-E). TF1-hMPL cells expressing shRNA to the DUB subunit of the BRISC complex, BRCC36, showed enhanced K63-polyUb of JAK2, which was abrogated by inhibitors to Ubc13, the E2 enzyme for K63Ub. In contrast, we failed to detect JAK2 K48Ub using K48Ub-TUBE, whereas Cyclin B1 K48Ub was detected as a positive control. Importantly, all BRISC-deficient cell lines (shBrcc36, shKiaa0157, and shMerit40) demonstrated an increased K63Ub signal compared with shRNA-luciferase controls. Proteasome inhibitors MG132 increased total ubiquitination levels in the cell as shown in the “input”; however, it did not increase K63-ubiquitination of JAK2 or pJAK2 at 10 minutes, indicating the specificity of the K63Ub-TUBE. It also reinforces the notion that JAK2 is largely K63-ubitiquitinated over K48-ubiquitinated at the early time point after TPO stimulation. Furthermore, the K63Ub signal was quickly attenuated by 20 minutes in the absence of MG132, whereas the signal was prolonged in the presence of MG132, peaking at 20 minutes after TPO stimulation, which is consistent with our recent report showing that phosphorylated JAK2 is ubiquitinated and degraded.26 Taken together, we showed that BRISC depletion increases TPO-induced JAK2 K63-polyUb.
BRISC depletion enhances hematopoietic cell growth and increases JAK2 protein levels as well as JAK2 activation
We previously showed that MERIT40 functions through attenuating TPO/Mpl signaling in HSCs.9 We therefore investigated the cellular response to TPO in BRISC-depleted hematopoietic cells. We plated TF-1 cells stably depleted of Luc, BRCC36, or KIAA in a graded concentration of Eltrombopag, a small molecule MPL agonist approved by the US Food and Drug Administration, or TPO. BRISC-depleted cells displayed a growth advantage in response to TPO agonists and TPO over Luc controls (Figure 4A-B).
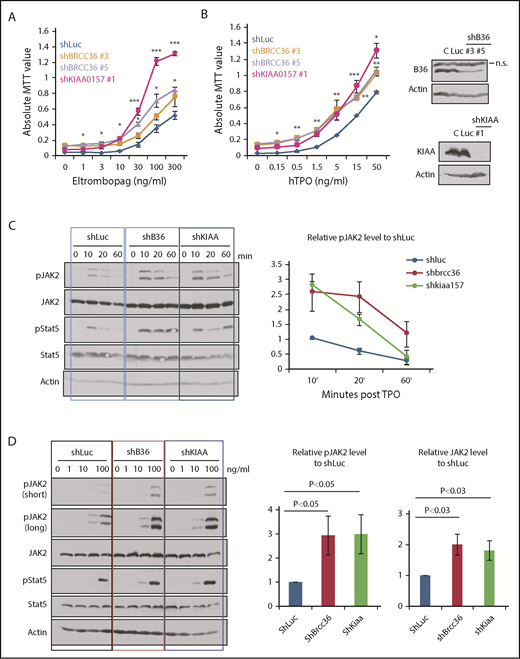
BRISC depletion enhances hematopoietic cell growth and increases JAK2 protein levels as well as JAK2 activation. (A-B) TF-1/hMPL cells stably expressing shRNAs to KIAA0157 or BRCC36 were established. Cells were cultured in different concentrations of MPL agonist Eltrombopag (A) or TPO (B). Live cell numbers after 3 days’ culture were determined by 3-(4,5-dimethylthiazole-2-yl)-2, 5-diphenyl tetrazolium bromide (MTT) absorbance. *P < .05; **P < .01; ***P < .001, 2 -tailed Student t test, shKIAA or shBRCC36 vs shRNA-luciferase (shLuc). (B, right panels) WB analysis shows efficient knockdown of individual BRISC components. shKIAA efficiently reduced BRCC36 protein levels. (C-D) TF1/hMPL cells depleted of various BRISC components (BRCC36 or KIAA0157) were starved and stimulated with TPO for different time points (C) or a graded dose of TPO for 10 minutes (D). (C-D, left panels) WB analysis with indicated antibodies is shown. (C, right) Relative pJAK2 levels upon TPO simulation to the shLuc controls at the 10-minute time point were quantified and plotted in the right panels. (D, right) Relative pJAK2 and total JAK2 protein levels to shLuc controls are quantified and plotted. N = 7. P values are determined by using the 2-tailed Student t test.
BRISC depletion enhances hematopoietic cell growth and increases JAK2 protein levels as well as JAK2 activation. (A-B) TF-1/hMPL cells stably expressing shRNAs to KIAA0157 or BRCC36 were established. Cells were cultured in different concentrations of MPL agonist Eltrombopag (A) or TPO (B). Live cell numbers after 3 days’ culture were determined by 3-(4,5-dimethylthiazole-2-yl)-2, 5-diphenyl tetrazolium bromide (MTT) absorbance. *P < .05; **P < .01; ***P < .001, 2 -tailed Student t test, shKIAA or shBRCC36 vs shRNA-luciferase (shLuc). (B, right panels) WB analysis shows efficient knockdown of individual BRISC components. shKIAA efficiently reduced BRCC36 protein levels. (C-D) TF1/hMPL cells depleted of various BRISC components (BRCC36 or KIAA0157) were starved and stimulated with TPO for different time points (C) or a graded dose of TPO for 10 minutes (D). (C-D, left panels) WB analysis with indicated antibodies is shown. (C, right) Relative pJAK2 levels upon TPO simulation to the shLuc controls at the 10-minute time point were quantified and plotted in the right panels. (D, right) Relative pJAK2 and total JAK2 protein levels to shLuc controls are quantified and plotted. N = 7. P values are determined by using the 2-tailed Student t test.
To further explore the signaling effects of enhanced K63-polyUb of JAK2 upon BRISC loss, we examined JAK2 activation kinetics and protein levels upon TPO stimulation. We found that BRISC depletion enhanced JAK2 activation approximately threefold and increased JAK2 protein level approximately twofold, indicating increased TPO sensitivity (Figure 4C-D). Therefore, BRISC deficiency potentiates JAK2 activation and the cellular responses to TPO.
LNK does not affect BRISC DUB activities
To examine the functional relationship between LNK and BRISC, we tested if LNK affects BRISC DUB activity. Because BRCC36 requires interaction with the other BRISC components for stability and enzymatic activity,23,25 we reconstituted all 4 BRISC components along with LNK, then subjected them to in vitro DUB assay. Cleaved Ubs were detected with or without LNK (supplemental Figure 4), suggesting that LNK does not affect BRISC DUB activity.
BRISC depletion enhances MPL surface levels as well as MPL-associated membrane proximal JAK2
K63-polyUb is known to regulate complex formation, receptor endocytic trafficking, autophagy, kinase activation, and DNA repair rather than proteasomal degradation.12-16 To our knowledge, this report is the first showing that a K63 DUB modulates JAK2 protein level or its activation. To investigate the mechanisms underlying this novel regulation, we first examined JAK2 half-life as well as MPL receptor endocytosis. We found that JAK2 was more stable and showed an enhanced activation in the absence of BRISC (Figure 5A). Furthermore, BRISC depletion increased total and cell surface levels of MPL, albeit the MPL receptor endocytosis rate remained unchanged (Figure 5A-B). We next assessed membrane-proximal JAK2 that is associated with cell surface MPL receptor, using surface-IP with anti-MPL antibodies in intact cells. After extensive washing, we permeabilized the cells and subsequently pulled down membrane MPL–associated JAK2 and pJAK2 visualized by WB analysis (Figure 5C-D). TPO stimulated an increased association between surface MPL and JAK2, and, more strikingly, an increased membrane association between MPL and pJAK2. BRCC36 depletion increased membrane proximal JAK2 and pJAK2 at the basal level, as well as 10 to 30 minutes upon TPO stimulation. Together, these data indicate that BRISC depletion increases JAK2 association with cell surface MPL, thus stabilizing JAK2 and enhancing JAK2 activation and signaling response promptly upon TPO stimulation.
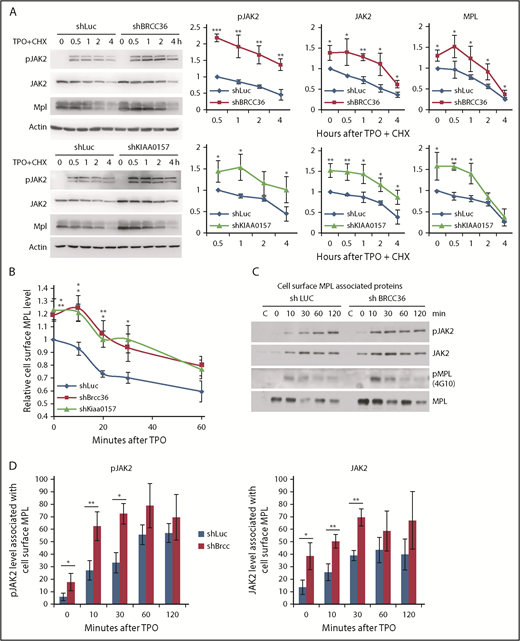
BRISC depletion enhances MPL surface levels as well as MPL-associated membrane proximal JAK2. (A) TF-1/MPL cells with stable depletion of BRCC36 or KIAA0157 were starved and stimulated with or without human TPO in the presence of cycloheximide (CHX) for indicated times. JAK2, pJAK2, and MPL half-lives were determined by WB analysis. Representative blots of 3 independent experiments are shown on the left. Relative protein levels to shRNA-luciferase (shLuc) controls at time 0 for JAK2 and MPL, or to time 0.5 hour for pJAK2, are shown on the right. (B) TF-1/MPL cell lines stably expressing shRNAs to Luc, BRCC36, or KIAA0157 were starved and stimulated with TPO for different times. Quantifications of TPO-stimulated MPL internalization relative to time 0 of the shLuc controls are shown. (C) JAK2 binds MPL upon ligand stimulation. TF1 cells stably expressing shRNAs to Luc or BRCC36 were starved for 3 hours and treated with TPO for indicated times. Cell surface MPL receptors were precipitated with anti-CD110 antibodies in native PBS buffer. Subsequently, cells were extensively washed and permeabilized with NP-40 and subjected to pulldown with Protein G agarose. Precipitates were then blotted with antibodies to pJAK2, JAK2, 4G10 for pMPL, and total MPL. The pulldowns were performed 3 to 5 times, and representative blots are shown. (D) Quantifications of TPO-stimulated pJAK2 and JAK2 associated with cell surface MPL receptor are shown. *P < .05; **P < .01; ***P < .001, 2-tailed Student t test.
BRISC depletion enhances MPL surface levels as well as MPL-associated membrane proximal JAK2. (A) TF-1/MPL cells with stable depletion of BRCC36 or KIAA0157 were starved and stimulated with or without human TPO in the presence of cycloheximide (CHX) for indicated times. JAK2, pJAK2, and MPL half-lives were determined by WB analysis. Representative blots of 3 independent experiments are shown on the left. Relative protein levels to shRNA-luciferase (shLuc) controls at time 0 for JAK2 and MPL, or to time 0.5 hour for pJAK2, are shown on the right. (B) TF-1/MPL cell lines stably expressing shRNAs to Luc, BRCC36, or KIAA0157 were starved and stimulated with TPO for different times. Quantifications of TPO-stimulated MPL internalization relative to time 0 of the shLuc controls are shown. (C) JAK2 binds MPL upon ligand stimulation. TF1 cells stably expressing shRNAs to Luc or BRCC36 were starved for 3 hours and treated with TPO for indicated times. Cell surface MPL receptors were precipitated with anti-CD110 antibodies in native PBS buffer. Subsequently, cells were extensively washed and permeabilized with NP-40 and subjected to pulldown with Protein G agarose. Precipitates were then blotted with antibodies to pJAK2, JAK2, 4G10 for pMPL, and total MPL. The pulldowns were performed 3 to 5 times, and representative blots are shown. (D) Quantifications of TPO-stimulated pJAK2 and JAK2 associated with cell surface MPL receptor are shown. *P < .05; **P < .01; ***P < .001, 2-tailed Student t test.
Discussion
Our findings, for the first time, implicate K63-polyUb–specific DUB activity as an important regulator of JAK2. BRISC regulates JAK2 activation and growth responses via removal of JAK2 K63-ubiquitination. TPO stimulation transiently increases JAK2 K63-polyUb in a manner that correlates with JAK2 phosphorylation and activation. BRISC deficiency increases JAK2 protein levels and potentiates JAK2 activation, thereby augmenting cellular responses to TPO and expansion of HSCs. Our findings suggest that BRISC controls membrane proximal JAK2 and pJAK2 coupled to cytokine receptor–mediated activation by K63-ubiquitination. Hence, this research identifies a novel mechanism of JAK2 stabilization via K63-polyUb that occurs independent of the proteasome.
BRISC is a 4-component protein complex that was first biochemically purified as a major K63-DUB activity in the cytoplasm.23 MERIT40 is required for the complex stability and DUB activity for both BRISC and nuclear BRCA1-A complexes.12,36,37 KIAA0157, which is unique to the BRISC complex, is essential for BRISC DUB activity in vitro and in vivo.23,25 BRISC deficiency by deleting Kiaa0157 increases the number of phenotypic and functional HSCs. Kiaa0157−/− mice exhibit phenotypes similar to those of M40−/− mice. Of note, Kiaa deficiency in BM showed a marked reduction in BRCC36, similarly to M40 deficiency, suggesting that BRISC is the major BRCC36-containing DUB complex in hematopoietic cells. This finding is also consistent with the observation that although M40−/− mice and cells are moderately sensitive to DNA damage,33 Kiaa0157−/− mice exhibit normal DNA damage response as well as increased HSC numbers.9
We present here a novel mechanism by which K63-ubiquitination regulates protein stability and kinase activation without affecting proteasome degradation. TPO stimulates an increased association between surface MPL and JAK2, and, more apparently, an increased membrane association between MPL and pJAK2. This scenario is in agreement with a recent report on a close MPL family member, the receptor for erythropoietin.38 Single molecular microscopy at the cell membrane in live cells showed that erythropoietin induces erythropoietin receptor dimerization that is required for full activation of JAK2. We showed that JAK2 activation correlates with a prompt induction of JAK2 K63-polyUb that is not affected by proteasome inhibitors. K63-polyUb normally promotes receptor endocytosis; however, we found that MPL surface level is increased at the basal state, but the endocytosis kinetics remain unchanged. Rather, our data indicate that K63-polyUb in JAK2 promotes membrane localization and association of JAK2 and pJAK2 with the MPL receptor, thereby promoting JAK2 stability and activation. However, the exact mechanism of how increased JAK2 K63-polyUb affects its association with membrane MPL has yet to be fully established.
We recently discovered that the CBL family E3 ubiquitin ligases promote JAK2 K63-ubiquitination and degradation.26 Cells depleted of CBL/CBLB showed a near complete abrogation of JAK2 K63-polyUb. It is counterintuitive that knockout of an E3 ligase and a DUB both result in increased JAK2 stability and HSC expansion. This research shows that CBL and BRISC control different aspects of JAK2 ubiquitination. Increased K63-ubiquitination upon BRISC loss promotes JAK2 stability and activation in a proteasome-independent manner, which is distinctly different from how CBL loss influences JAK2 activity. The overall stability of JAK2 and pJAK2 activation kinetics are largely regulated by CBL, whereas BRISC effects are relatively transient, moderate, and are at the membrane receptor level. As a result, CBL has a much stronger effect on JAK2 stability and signaling than that of BRISC, loss of which exhibits a marked HSC expansion that ultimately contributes to development of myeloid leukemia.26 Dissecting the different Ub chain modifications and their respective roles in JAK2 signaling warrants future investigation.
Interestingly, mutations in BRCC36 (also called BRCC3) are found in human clonal hematopoiesis.39 Our studies shed light on the mechanisms by which BRCC36 DUB regulates HSC expansion and hematopoietic dysregulation. Moreover, our research highlights the complex relationship between ubiquitination on kinase activation and its impact on stem cell maintenance. Because DUBs are specialized proteases that are deemed potential “druggable” targets, this research holds promise that BRISC-specific inhibition may represent an approach to increase the efficacy of stem cell transplantation.
Protocol and reagents generated in this work will be made available upon request.
The online version of the article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
W.T. was supported by grants from the National Institutes of Health, National Heart, Lung, and Blood Institute (R01HL095675 and R01 HL133828), the Department of Defense, the Basser Center for BRCA Team Science Grant, and awards from the Fanconi Anemia Research Fund and Leukemia Lymphoma Society. R.A.G. is supported by a grant from the National Institutes of Health, National Cancer Institute (R01CA138835) and the Basser Center for BRCA.
Authorship
Contribution: W.T., R.D., K.R., K.L., and R.A.G. designed experiments, interpreted results, and wrote the manuscript; R.D., X.H., K.R., Q.J., K.L., and M.D. performed the experiments and analyzed the data; and R.A.G. generated Kiaa0157−/− mice and edited the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Wei Tong, Children’s Hospital of Philadelphia, Abramson Building, Suite 310D, 3615 Civic Center Blvd, Philadelphia, PA 19104-4318; e-mail: tongw@email.chop.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal