

Key Points
IDH2 is a new synthetic lethal target to PI, efficacious in several hematological malignancies.
Inhibition of NAMPT/SIRT3/IDH2 pathway could enhance the therapeutic efficacy and overcome resistance to PI.

Abstract
Proteasome inhibitors (PI) are extensively used for the therapy of multiple myeloma (MM) and mantle cell lymphoma. However, patients continuously relapse or are intrinsically resistant to this class of drugs. Here, to identify targets that synergize with PI, we carried out a functional screening in MM cell lines using a short hairpin RNA library against cancer driver genes. Isocitrate dehydrogenase 2 (IDH2) was identified as a top candidate, showing a synthetic lethal activity with the PI carfilzomib (CFZ). Combinations of US Food and Drug Administration–approved PI with a pharmacological IDH2 inhibitor (AGI-6780) triggered synergistic cytotoxicity in MM, mantle cell lymphoma, and Burkitt lymphoma cell lines. CFZ/AGI-6780 treatment increased death of primary CD138+ cells from MM patients and exhibited a favorable cytotoxicity profile toward peripheral blood mononuclear cells and bone marrow–derived stromal cells. Mechanistically, the CFZ/AGI-6780 combination significantly decreased tricarboxylic acid cycle activity and adenosine triphosphate levels as a consequence of enhanced IDH2 enzymatic inhibition. Specifically, CFZ treatment reduced the expression of nicotinamide phosphoribosyltransferase (NAMPT), thus limiting IDH2 activation through the NAD+-dependent deacetylase SIRT3. Consistently, combination of CFZ with either NAMPT or SIRT3 inhibitors impaired IDH2 activity and increased MM cell death. Finally, inducible IDH2 knockdown enhanced the therapeutic efficacy of CFZ in a subcutaneous xenograft model of MM, resulting in inhibition of tumor progression and extended survival. Taken together, these findings indicate that NAMPT/SIRT3/IDH2 pathway inhibition enhances the therapeutic efficacy of PI, thus providing compelling evidence for treatments with lower and less toxic doses and broadening the application of PI to other malignancies.
Introduction
The ubiquitin-proteasome pathway plays a crucial role in protein processing and degradation, regulating critical cellular functions including cell-cycle control, transcriptional regulation, cellular stress responses, and antigen presentation.1 It is well established that proteasome inhibition results in the disruption of normal homeostatic mechanisms, and that malignant cells are more susceptible to the cytotoxic effects of proteasome inhibition than normal cells, most likely as a consequence of their increased requirement for protein synthesis and their higher levels of proteasome activity.2 A number of processes have been reported to contribute to the antitumoral effects of proteasome inhibitors (PI), including inhibition of the NF-κB pathway,3 altered cell-cycle control and apoptosis mechanisms,4,5 endoplasmic reticulum stress, suppression of cell adhesion signaling, inhibition of angiogenesis, and DNA repair.2 The prevalent sensitivity of transformed cells to PI and the successful design of clinical protocols have led to the regulatory approval of PI to treat multiple myeloma (MM) and mantle cell lymphoma (MCL) patients.6-10 To date, 3 PI are routinely used in clinical settings (bortezomib [BTZ], carfilzomib [CFZ], and ixazomib), and additional PI are under investigation.11 The pleiotropic consequences of proteasome inhibition result in synergistic or additive activity with other therapeutic protocols, including autologous stem cell transplantation, glucocorticoids, alkylating agents and anthracyclines, immunomodulatory drugs, histone deacetylase inhibitors, and monoclonal antibodies.10,12 Despite these enormous advances, relapses and disease progressions are common among MM patients, suggesting a prominent role for either innate or acquired drug resistance.13,14 Moreover, although the toxicity of PI is quite well controlled in clinical settings, they display distinct adverse profiles, imposing limits to their doses.15 In addition, responses to PI in other hematological malignancies have been contradictory.6,16,17 Similarly, promising preclinical data obtained with PI in models of solid tumors have not been confirmed in the clinic,15 probably as a consequence of impaired drug distribution, requiring higher dosages, not applicable for the toxic effects. Therefore, the design of a new generation of ubiquitin-proteasome pathway inhibitors and the identification of novel combination strategies is essential to overcome resistance and broaden the applicability of this class of drugs to other hematological malignancies, and possibly to solid tumors.
Here, to identify druggable targets that inhibition sensitizes MM cells to PI, we performed a short hairpin RNA (shRNA) functional screening targeting 152 cancer driver genes. Isocitrate dehydrogenase 2 (IDH2) silencing revealed synthetic lethal activity with CFZ. Combinations of the pharmacological IDH2 inhibitor AGI-6780 with PI triggered synergistic cytotoxicity in MM, MCL, and Burkitt lymphoma (BL) cell lines, as well as in primary CD138+ cells from MM patients. Importantly, inducible IDH2 knock-down enhanced the therapeutic efficacy of CFZ in a subcutaneous xenograft model of MM. Our findings indicate that the nicotinamide phosphoribosyltransferase (NAMPT)/SIRT3/IDH2 pathway is a major determinant of PI responsiveness in hematological malignancies, thus providing proof of concept for new combination strategies to enhance sensitivity and overcome resistance to PI.
Materials and methods
Detailed experimental procedures for cell culture conditions, shRNA screening, plasmid constructs, virus production, in vitro transduction, generation of inducible cell lines, purification of total RNA and reverse transcription-quantitative polymerase chain reaction, DNA sequencing, western blotting, gene expression profiling, analysis of apoptosis and cell cycle, analysis of reactive oxygen species (ROS) production, mitochondria isolation, and NF-κB activity are included in supplemental Material and methods, available on the Blood Web site.
TCA cycle measurement
The glucose flux through tricarboxylic acid (TCA) cycle was measured by radiolabeling cells with 2 μCi/mL [6-14C]-glucose (55 mCi/mmol; PerkinElmer, Waltham, MA). Cell suspensions were incubated for 1 hour in a closed experimental system to trap the 14CO2 developed from [14C]-glucose; the reaction was stopped by injecting 0.5 mL of 0.8 N HClO4. The amount of glucose transformed into CO2 through the TCA cycle was calculated as described previously,18 and expressed as picomoles CO2/h per milligram cell proteins.
IDH enzymatic activity
IDH activity was measured using the IDH assay kit (Sigma-Aldrich, St Louis, MO), according to the manufacturer’s protocol. IDH activity was determined using isocitrate as a substrate of the reaction, which results in a colorimetric (450 nm) product proportional to the enzymatic activity present. One unit of IDH is the amount of enzyme that generates 1.0 μmole of NADH or NAD phosphate (NADP) per minute at pH 8.0 at 37°C. To evaluate IDH2 and IDH1 activities, mitochondrial or cytoplasmic extracts were used, respectively.
Measurement of complex I-III activity
Mitochondria were extracted as described in the supplemental Materials and methods. The electron flux from complex I to complex III was measured in 50 μL nonsonicated mitochondrial extracts, resuspended in 120 μL buffer A (5 mM KH2PO4, 5 mM MgCl2, 5% w/v bovine serum albumin) in a 96-well plate. Then, 100 μL buffer B (25% w/v saponin, 50 mM KH2PO4, 5 mM MgCl2, 5% w/v bovine serum albumin, 0.12 mM cytochrome c-oxidized form, and 0.2 mM NaN3) was added for 5 minutes at room temperature. The reaction was started with 0.15 mM NADH and was followed for 6 minutes, reading the absorbance at 550 nm by a Λ 3 spectrophotometer (PerkinElmer). Results were expressed as nanomoles cytochrome c reduced/min milligram mitochondrial proteins.19
ATP measurement
The amount of adenosine triphosphate (ATP) was measured in 50 μL mitochondrial extracts with the ATPlite assay (PerkinElmer), using a Synergy HT Multi-Mode Microplate Reader (Bio-Tek Instruments, Winooski, VT). ATP was quantified as arbitrary light units; data were converted into nanomoles per milligram mitochondrial proteins, using a calibration curve previously set.
Xenograft models
KMS-27-tTR-KRAB (TK) IDH2-A4 cells (5 × 105) suspended in phosphate-buffered saline–50% Matrigel (BD Biosciences, San Jose, CA) were injected into the left and right flanks of NOD/SCID/IL2Rγ−/− (NSG) mice, previously anesthetized intramuscularly with xylazine and tiletamine/zolazepam. Tumor growth was monitored over time by determining the volume of tumor masses. Mice with tumor masses of 0.5 cm diameter (∼3 weeks after the injection) were randomized and treated for 3 weeks with doxycycline (DOXY) by oral administration (0.1 mg/mL biweekly), CFZ IV (4 mg/kg biweekly), or the combination with the same dosing regimen used for the individual agents. DOXY was administrated in a 0.5% sucrose solution in light-proof bottles, for 48 hours. CFZ was dissolved in 3% dimethyl sulfoxide (DMSO), 10% Captisol (CYDEX Pharmaceuticals Inc., Lenexa, KS), 10 mM sodium citrate pH 3.5, and administrated after DOXY removal. The control group received the carriers alone at the same schedule as the combination group. Mice were euthanized in a carbon dioxide chamber after the tumor masses reached a volume of approximately 1500 mm3 or at early signs of distress. Tumor volume was calculated using the ellipsoid equation 4/3 × π × 1/2 × (length × width × depth). Animals were housed in the animal facility of the Molecular Biotechnology Center (Torino, Italy), in accordance with guidelines approved by the local Ethical Animal Committee. Experimental approval was obtained from the Italian Ministry of Health.
Statistical analysis
Statistical analyses were performed with GraphPad Prism 5.01 (GraphPad Software Inc.). Statistical significance of differences observed (in both in vitro and in vivo experiments) was determined by Student t test; differences were considered significant when P value was <.05 (*), <.01 (**), or <.001 (***). Survival curves were estimated with the Kaplan-Meier method. The log-rank test was used for statistical analysis.
Results
shRNA screening in multiple myeloma cell lines identifies IDH2 gene as synthetic lethal to the proteasome inhibitor carfilzomib
To identify druggable targets that synergize with PI, we generated 2 MM cell lines (KMM-1PIR and U266PIR) cross-resistant to the PI BTZ and CFZ (supplemental Figure 1). A functional screening using a shRNA library targeting 152 cancer driver genes, highly representative of all signaling pathways, was carried out in the KMM-1PIR cell line treated with sublethal concentrations of CFZ (Figure 1A-B; supplemental Tables 1-6). The primary screening was validated in the U266PIR cell line by targeting the top 24 genes (supplemental Table 7). Analysis of the correlation between gene silencing efficacy and growth inhibition in presence of CFZ led to the identification of 3 synthetic lethal target genes (Figure 1C). Further studies were focused on IDH2, an NADP+ dependent mitochondrial enzyme that catalyzes the oxidative decarboxylation of isocitrate to α-ketoglutarate in the TCA cycle. To validate screening results, 2 shRNA sequences (A4 and A6) directed against human IDH2 were individually transduced into KMM-1PIR and U266PIR cells (supplemental Figure 2). IDH2 knockdown did not affect viability of KMM-1PIR and U266PIR cells. In contrast, IDH2 depletion was dramatically cytotoxic in cells treated with a sublethal dose of CFZ (Figure 1D-E). We excluded that IDH2 mutations or its aberrant expression were associated to PI resistance in MM cells (supplemental Figure 3). These findings prompted us to verify whether IDH2 knockdown could synergize with CFZ also in PI-sensitive cell lines. Accordingly, IDH2 silencing considerably enhanced sensitivity to CFZ in parental KMM-1 and U266 cell lines (Figure 1F-G). Taken together, these data established that IDH2 knockdown is synthetic lethal to CFZ treatment in both PI-resistant and PI-sensitive MM cell lines.

shRNA screening in MM cell lines identifies IDH2 gene as synthetic lethal to the proteasome inhibitor carfilzomib. (A) Experimental design of the shRNA screen to identify genes conferring sensitivity to CFZ in MM cells. KMM-1PIR cells were infected with 684 shRNAs targeting 152 cancer driver genes (day −3) and incubated in presence or absence of puromycin (day −2). KMM-1PIR cells were then split and treated with 2.5 nM CFZ or with control diluent (DMSO) (day 0). Growth rate was calculated at day 3 and 7 posttreatment (supplemental Table 5), and positive hits selected according to the z score. Top 24 selected genes were validated in a secondary screening performed in U266PIR cells. (B) Representation of the z score (y-axis) for every shRNA (x-axis) calculated on growth rate reduction for each shRNA. Red box highlights candidates with z score below −0.8 (day 7) (supplemental Table 6). (C) Correlation between percentage of gene silencing and percentage of growth inhibition in presence of CFZ for top 3 candidate genes (IDH2, KDM1A, and SOX2) in U266PIR cells. (D) KMM-1PIR, (E) U266PIR, (F) KMM-1, and (G) U266 cell lines were transduced with the empty vector or shRNAs targeting IDH2 (shIDH2_A4, shIDH2_A6) and treated with CFZ (KMM-1PIR and U266PIR, 5 nM; KMM-1 and U266, 2.5 nM) or DMSO every 48 hours. Cell viability was measured by TMRM staining-flow cytometry 96 hours posttreatment for KMM-1PIR and U266PIR and 48 hours posttreatment for KMM-1 and U266. Data are the means ± standard deviation (SD) of 3 independent experiments (*P < .05; **P < .01). TMRM, tetramethylrhodamine.
shRNA screening in MM cell lines identifies IDH2 gene as synthetic lethal to the proteasome inhibitor carfilzomib. (A) Experimental design of the shRNA screen to identify genes conferring sensitivity to CFZ in MM cells. KMM-1PIR cells were infected with 684 shRNAs targeting 152 cancer driver genes (day −3) and incubated in presence or absence of puromycin (day −2). KMM-1PIR cells were then split and treated with 2.5 nM CFZ or with control diluent (DMSO) (day 0). Growth rate was calculated at day 3 and 7 posttreatment (supplemental Table 5), and positive hits selected according to the z score. Top 24 selected genes were validated in a secondary screening performed in U266PIR cells. (B) Representation of the z score (y-axis) for every shRNA (x-axis) calculated on growth rate reduction for each shRNA. Red box highlights candidates with z score below −0.8 (day 7) (supplemental Table 6). (C) Correlation between percentage of gene silencing and percentage of growth inhibition in presence of CFZ for top 3 candidate genes (IDH2, KDM1A, and SOX2) in U266PIR cells. (D) KMM-1PIR, (E) U266PIR, (F) KMM-1, and (G) U266 cell lines were transduced with the empty vector or shRNAs targeting IDH2 (shIDH2_A4, shIDH2_A6) and treated with CFZ (KMM-1PIR and U266PIR, 5 nM; KMM-1 and U266, 2.5 nM) or DMSO every 48 hours. Cell viability was measured by TMRM staining-flow cytometry 96 hours posttreatment for KMM-1PIR and U266PIR and 48 hours posttreatment for KMM-1 and U266. Data are the means ± standard deviation (SD) of 3 independent experiments (*P < .05; **P < .01). TMRM, tetramethylrhodamine.
Pharmacological inhibition of IDH2 enhances sensitivity to CFZ in MM cell lines
To define whether pharmacological inhibition of IDH2 recapitulates the synthetic lethal phenotype, CFZ treatment was associated to AGI-6780, an allosteric inhibitor of mutant IDH2 that is known to reduce the activity of wild-type IDH2, although less potently.20,21 We first demonstrated that AGI-6780 (5 μM) selectively impaired IDH2 enzymatic activity in MM cells (supplemental Figure 4A-D). Next, the PI-resistant MM cell lines KMM-1PIR and U266PIR were treated with either CFZ, AGI-6780, or a combination of the 2 drugs. Combinatorial treatments significantly increased cell death, compared with single drugs (Figure 2A-B), confirming data obtained by IDH2 knockdown. This combination was effective also in MM cells resistant to very high concentrations of PI (Figure 2C; supplemental Figure 5). To prove that the combined cytotoxicity of AGI-6780 and CFZ is not restricted to PI-resistant cells, 8 MM cell lines with different degrees of PI sensitivity were treated with a single dose of CFZ in combination or not with AGI-6780, refreshed every 48 hours. Enhanced sensitivity to the combination treatment in comparison with either agent alone was observed in all MM cell lines (Figure 2D; supplemental Figure 6A-B). In contrast, the chronic myelogenous leukemia cell line K-562 was unresponsive to both drugs and to their combination (Figure 2D). Increased sensitivity to CFZ was confirmed in 2 MM cell lines (KMS-27 and U266) by regimens with lower doses of CFZ administered every 48 hours in combination with AGI-6780 (supplemental Figure 6C-D). Considering that hypoxic bone marrow microenvironment favors MM progression and drug resistance, we tested if this environment could affect the response to PI and AGI-6780.22-24 We confirmed that the combination of the 2 drugs increased MM cell death, also in presence of cells cultured with 1% oxygen concentration (supplemental Figure 7). To elucidate mechanisms of synthetic lethality, cell-cycle and apoptotic markers were analyzed. CFZ/AGI-6780 combination was associated with an increase of G0/G1 phase (supplemental Figure 8), down-modulation of cyclins, upregulation of cyclin-dependent kinase inhibitors, proteolytic cleavage of the caspases substrate PARP-1, and activation of effector caspases 3, 7, and 9 (Figure 2E). To reduce the confounding effects of cell death induction, western blotting and cell-cycle analysis were performed 24 hours posttreatment, when cells displayed comparable levels of viability (Figure 2F-G). To further define the molecular mechanisms involved and/or regulated by the synergistic activity of CFZ/AGI-6780, gene expression profiles were analyzed 6 and 12 hours after single or combination treatments and compared with untreated control samples. Supervised analysis identified 115 genes differentially regulated by CFZ, whereas AGI-6780 treatment had negligible transcriptional effects. Remarkably, 261 genes were differentially expressed after combined treatment, and nearly all genes modulated by CFZ (106/115) were concordantly modified to a higher degree by CFZ/AGI-6780 treatment (supplemental Figure 9A). Pathway analyses confirmed that the classical targets of PI such as unfolded protein response, NF-κB, cell cycle, and apoptosis, were affected in response to CFZ alone; these effects were enhanced by the combination with AGI-6780 (supplemental Figure 9B). Collectively, these findings indicate that the CFZ/AGI-6780 regimen is effective against PI-resistant and PI-sensitive MM cells and elicits significant changes converging in cell cycle and apoptotic pathways.
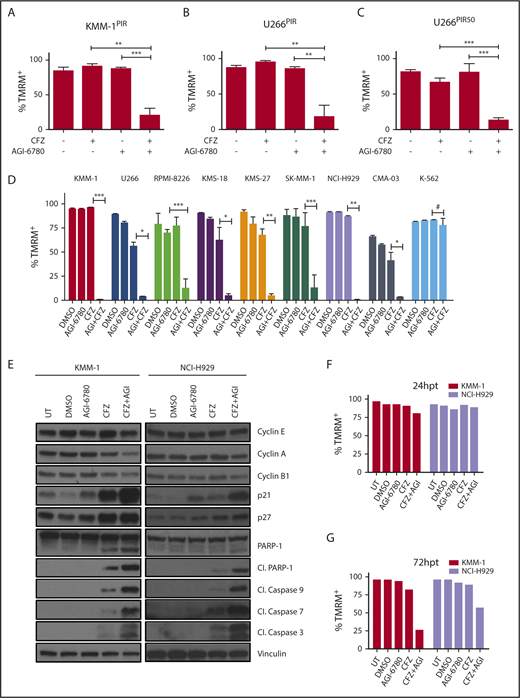
Pharmacological inhibition of IDH2 enhances sensitivity to CFZ in MM cell lines. (A) KMM-1PIR and (B) U266PIR cells were treated with 2.5 nM CFZ in combination or not with 10 μM AGI-6780. Cell viability was measured by TMRM staining-flow cytometry 96 hours posttreatment. Data are the means ± SD of 4 independent experiments. (C) U266PIR50 cells were treated with 75 nM CFZ in combination or not with 10 μM AGI-6780. Cell viability was measured by TMRM staining-flow cytometry 72 hours posttreatment. Data are the means ± SD of 4 independent experiments. (D) Eight MM cell lines and the K-562 cell line were treated with CFZ (1.67 nM CFZ for KMS-18; 2.5 nM for RPMI-8226, KMS-27, SK-MM-1, and CMA-03; 5 nM for KMM-1, U266, and NCI-H929 cell lines) in combination or not with 5 μM AGI-6780 (2.5 μM for RPMI-8226). Treatment was performed every 48 hours for AGI-6780, but only at day 0 for CFZ. Cell viability was measured by TMRM staining-flow cytometry 8 days posttreatment. Data are the means ± SD of 3 independent experiments (*P < .05; **P < .01; ***P < .001; #P ≥ .05). (E) Western blot of KMM-1 and NCI-H929 cells, UT, treated with DMSO, AGI-6780 (KMM-1: 5 μM; NCI-H929: 10 μM), CFZ (KMM-1: 5 nM; NCI-H929: 2.5 nM), or a combination of the 2 drugs. Cell lysates were immunoblotted using the indicated antibodies 24 hours posttreatment. Vinculin protein expression was included for protein loading normalization. (F-G) Cell viability of the experiment described previously was measured by TMRM staining-flow cytometry 24 and 72 hpt, respectively. Cl., cleaved; hpt, hours posttreatment; UT, untreated.
Pharmacological inhibition of IDH2 enhances sensitivity to CFZ in MM cell lines. (A) KMM-1PIR and (B) U266PIR cells were treated with 2.5 nM CFZ in combination or not with 10 μM AGI-6780. Cell viability was measured by TMRM staining-flow cytometry 96 hours posttreatment. Data are the means ± SD of 4 independent experiments. (C) U266PIR50 cells were treated with 75 nM CFZ in combination or not with 10 μM AGI-6780. Cell viability was measured by TMRM staining-flow cytometry 72 hours posttreatment. Data are the means ± SD of 4 independent experiments. (D) Eight MM cell lines and the K-562 cell line were treated with CFZ (1.67 nM CFZ for KMS-18; 2.5 nM for RPMI-8226, KMS-27, SK-MM-1, and CMA-03; 5 nM for KMM-1, U266, and NCI-H929 cell lines) in combination or not with 5 μM AGI-6780 (2.5 μM for RPMI-8226). Treatment was performed every 48 hours for AGI-6780, but only at day 0 for CFZ. Cell viability was measured by TMRM staining-flow cytometry 8 days posttreatment. Data are the means ± SD of 3 independent experiments (*P < .05; **P < .01; ***P < .001; #P ≥ .05). (E) Western blot of KMM-1 and NCI-H929 cells, UT, treated with DMSO, AGI-6780 (KMM-1: 5 μM; NCI-H929: 10 μM), CFZ (KMM-1: 5 nM; NCI-H929: 2.5 nM), or a combination of the 2 drugs. Cell lysates were immunoblotted using the indicated antibodies 24 hours posttreatment. Vinculin protein expression was included for protein loading normalization. (F-G) Cell viability of the experiment described previously was measured by TMRM staining-flow cytometry 24 and 72 hpt, respectively. Cl., cleaved; hpt, hours posttreatment; UT, untreated.
IDH2 inhibition synergizes with first- and second-generation PI in B-cell hematological malignancies
To expand the clinical relevance of our observations and demonstrate that IDH2 inhibition specifically synergize with PI, we first demonstrated that MM cells treated with AGI-6780 displayed enhanced response to the US Food and Drug Administration–approved PI BTZ and ixazomib (supplemental Figure 10A-B). Because PI have also been approved for the treatment of MCL patients and their anticancer effects have been obtained in different types of hematological malignancies,6,25 we tested whether IDH2 inhibition could synergize with PI in B-cell non-Hodgkin lymphoma models. Remarkably, a dramatic increase of cell death was observed in all MCL and BL cell lines treated with CFZ/AGI-6780 combinations (Figure 3). We then asked whether increased IDH2 activity could impair the cytotoxicity of PI. Because it is known that SIRT3 protein deacetylates IDH2 and enhances its activity under glucose deprivation,26,27 we cultured KMM-1 cells in absence of glucose for 7 days and measured IDH1, IDH2, and IDH3 enzymatic activities. As expected, a stable induction of IDH2 activity was observed after glucose restriction (supplemental Figure 10C). Next, we evaluated whether IDH2 activation was able to rescue MM cells from the effect of a CFZ/AGI-6780 combination. KMM-1 cells were conditioned by glucose deprivation for 24 hours and subsequently treated with CFZ, AGI-6780, or with both agents. Significantly, glucose restriction increased the viability of CFZ- and CFZ/AGI-6780–treated cells compared with not-starved cells (supplemental Figure 10D). Moreover, we performed a canonical rescue experiment overexpressing IDH2 and/or SIRT3 in KMM-1PIR cells (supplemental Figure 10E). We observed that only the combined overexpression of the 2 genes was able to enhance IDH2 activity (supplemental Figure 10F). Concordantly, cells with hyperactivation of IDH2 treated with CFZ and AGI-6780 partially decrease cell death, compared with the cells with a basal IDH2 activity (supplemental Figure 10G). Taken together, these results suggest that IDH2 activity antagonizes the therapeutic efficacy of first- and second-generation PI and that pharmacological IDH2 inhibition is a suitable strategy to enhance the therapeutic efficacy of PI in MM and other B-cell hematological malignancies.

IDH2 inhibition increases sensitivity to CFZ in mantle cell lymphoma and Burkitt lymphoma cells. (A) JeKo-1, (B) SP-49, (C) Mino, (D) Granta-519, (E) HS-Sultan, and (F) Raji cells were left UT, treated with DMSO, CFZ, AGI-6780, or a combination of the 2 drugs. JeKo-1 cells were treated at time 0, 48 hours, and 96 hours with both drugs. SP-49 cells were treated at time 0 and 48 hours with both drugs and at 96 hours with AGI-6780. Mino cells were treated with both drugs at time 0 and with AGI-6780 at 48 and 96 hours. Granta-519 cells were treated at time 0 and 48 hours with both drugs and every 48 hours with AGI-6780. HS-Sultan cells were treated at time 0 with both drugs and every 48 hours with AGI-6780. Raji cells were treated at time 0 and 48 hours with both drugs and every 48 hours with AGI-6780. Cell viability was measured by TMRM or annexin V staining-flow cytometry at the indicated time points. Data are the means ± SD of 4 independent experiments (*P < .05; **P <. 01; ***P <. 001). dpt, days posttreatment.
IDH2 inhibition increases sensitivity to CFZ in mantle cell lymphoma and Burkitt lymphoma cells. (A) JeKo-1, (B) SP-49, (C) Mino, (D) Granta-519, (E) HS-Sultan, and (F) Raji cells were left UT, treated with DMSO, CFZ, AGI-6780, or a combination of the 2 drugs. JeKo-1 cells were treated at time 0, 48 hours, and 96 hours with both drugs. SP-49 cells were treated at time 0 and 48 hours with both drugs and at 96 hours with AGI-6780. Mino cells were treated with both drugs at time 0 and with AGI-6780 at 48 and 96 hours. Granta-519 cells were treated at time 0 and 48 hours with both drugs and every 48 hours with AGI-6780. HS-Sultan cells were treated at time 0 with both drugs and every 48 hours with AGI-6780. Raji cells were treated at time 0 and 48 hours with both drugs and every 48 hours with AGI-6780. Cell viability was measured by TMRM or annexin V staining-flow cytometry at the indicated time points. Data are the means ± SD of 4 independent experiments (*P < .05; **P <. 01; ***P <. 001). dpt, days posttreatment.
CFZ/AGI-6780 combinatorial treatment decreases TCA cycle activity and mitochondrial ATP production through the NAMPT/SIRT3/IDH2 pathway
To define the molecular mechanisms responsible for the synergy between PI and IDH2 inhibition, we considered that targeting IDH2 activity could lead to a decrease of reduced NAD phosphate (NADPH) production, resulting in higher ROS levels.28 Taking into account that oxidative stress has been identified as an important mechanism of PI cytotoxicity in myeloma and nonmyeloma cells,29,30 we hypothesized that the CFZ/AGI-6780 combination could exacerbate ROS levels, leading to increased cell death. However, only a slight increase in mitochondrial ROS concentration was observed in MM cells treated with the CFZ/AGI-6780 combination (supplemental Figure 11). Next, we evaluated if IDH2 inhibition could impair TCA cycle activity.28 Notably, we observed that the CFZ/AGI-6780 combination more drastically decreased IDH2 and TCA cycle activities, despite CFZ treatment being ineffective (Figure 4A-D). In this setting, IDH2 inhibition was associated to a proportional increase in IDH1 and IDH3 activities (supplemental Figure 12). In addition, electron transport chain (ETC) flux and mitochondrial ATP synthesis were accordingly downregulated in MM cells treated with the combination of the 2 drugs (Figure 4E-F). Subsequently, we examined the biochemical mechanisms whereby CFZ treatment could synergize with AGI-6780 to further decrease IDH2 activity. It is recognized that PI inhibit NF-κB,10,31 and that expression of NAMPT, a rate-limiting enzyme in the NAD+ synthesis and sirtuins activation,32 is transcriptionally modulated by NF-κB.33-35 Therefore, we reasoned that PI could affect IDH2 activation through the NAMPT/NAD+/SIRT3 pathway (Figure 5A). Consistent with this hypothesis, we demonstrated that CFZ treatment significantly reduced NF-κB activity in KMS-27 cells (Figure 5B). Accordingly, NAMPT expression levels were significantly downregulated by CFZ treatment (Figure 5C). To confirm the involvement of the NAMPT/NAD+/SIRT3 pathway, we associated CFZ with several NAMPT inhibitors (FK866, GMX-1778, and Nampt-IN-1). As expected, combination of CFZ with NAMPT inhibitors induced synergistic downregulation of IDH2 and TCA activity (Figure 5D; supplemental Figure 13A), followed by MM cell death, confirming the synthetic lethality previously reported by Cagnetta et al with BTZ and FK866 (Figure 5E; supplemental Figure 13B-C).36 Importantly, these results were phenocopied by associating CFZ treatment to SIRT3 inhibition, both using specific drugs (AGK7 and TYP-3) (Figure 5F-G; supplemental Figure 13D-E) and shRNAs targeting SIRT3 (supplemental Figure 13F-G).37 Taken together, these data demonstrate that the CFZ/AGI-6780 combination significantly decreases TCA cycle activity as a consequence of enhanced IDH2 enzymatic inhibition. Specifically, CFZ treatment reduces NAMPT expression and thus limits IDH2 activation through the NAD+-dependent deacetylase SIRT3.
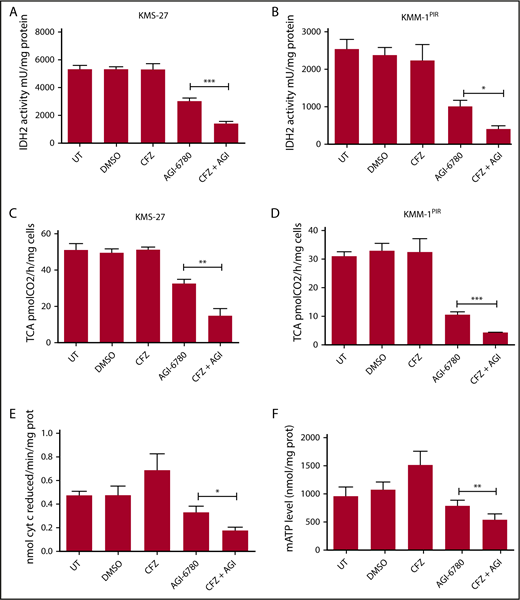
Combinatorial treatment with CFZ and AGI-6780 causes a reduction in IDH2 activity and mATP levels. (A) KMS-27 and (B) KMM-1PIR cells UT, treated with DMSO, CFZ (2.5 nM and 5 nM, respectively), AGI-6780 (5 µM), or a combination of the 2 drugs were analyzed for IDH2 activity 6 hours posttreatment. (C) KMS-27 and (D) KMM-1PIR cells treated as described previously were analyzed for TCA cycle activity 6 hours posttreatment. Data are the means ± SD of 4 independent experiments. (E) KMS-27 cells treated as described previously were analyzed for ETC complexes I to III 7 hours posttreatment. (F) KMS-27 cells treated as described previously were analyzed for mATP production 7 hours posttreatment. Data are the means ± SD of 3 independent experiments (*P < .05; **P < .01; ***P < .001). mATP, mitochondrial ATP.
Combinatorial treatment with CFZ and AGI-6780 causes a reduction in IDH2 activity and mATP levels. (A) KMS-27 and (B) KMM-1PIR cells UT, treated with DMSO, CFZ (2.5 nM and 5 nM, respectively), AGI-6780 (5 µM), or a combination of the 2 drugs were analyzed for IDH2 activity 6 hours posttreatment. (C) KMS-27 and (D) KMM-1PIR cells treated as described previously were analyzed for TCA cycle activity 6 hours posttreatment. Data are the means ± SD of 4 independent experiments. (E) KMS-27 cells treated as described previously were analyzed for ETC complexes I to III 7 hours posttreatment. (F) KMS-27 cells treated as described previously were analyzed for mATP production 7 hours posttreatment. Data are the means ± SD of 3 independent experiments (*P < .05; **P < .01; ***P < .001). mATP, mitochondrial ATP.
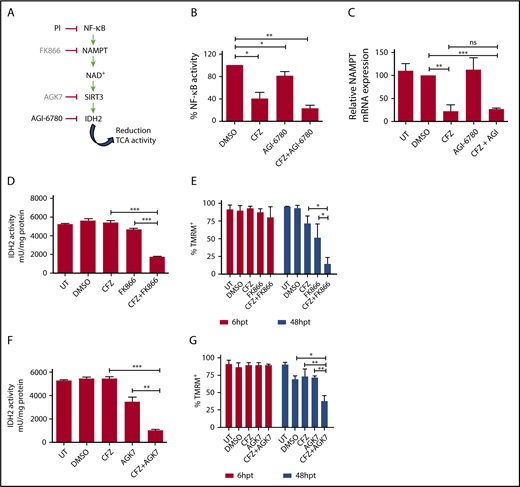
Combinatorial treatment with CFZ and AGI-6780 acts through the inhibition of the NAMPT/SIRT/IDH2 pathway. (A) Schematic representation of the NAMPT/SIRT3/IDH2 pathway and inhibitors. (B) KMS-27 cells treated with DMSO, AGI-6780 (5 µM), CFZ (3 nM), or a combination of the 2 drugs were analyzed for NF-κB activity 6 hours posttreatment. NF-κB activity was detected in total extracts measuring the DNA-binding capability of NF-κB on its target sequence (see “Materials and methods”). Data represent the percentage of NF-κB binding activity normalized vs DMSO samples and are the means ± SD of 3 independent experiments. (C) KMS-27 cells UT, treated with DMSO, CFZ (2.5 nM), AGI-6780 (5 µM), or a combination of the 2 drugs were analyzed for NAMPT mRNA expression levels 24 hours posttreatment. Data are the means ± SD of 3 independent experiments. (D-E) KMS-27 cells were left UT, treated with DMSO or FK866 (10 nM), for 48 hours; vehicle or CFZ (2.5 nM) were added for additional 48 hours. Cells were analyzed for (D) IDH2 activity 6 hours posttreatment with CFZ and for (E) cell viability by TMRM staining-flow cytometry 6 and 48 hpt with CFZ. Data are the means ± SD of 3 independent experiments. (F-G) KMS-27 cells UT, treated with DMSO, 1.25 nM CFZ, 10 µM AGK7, or a combination of the 2 drugs were analyzed for (F) IDH2 activity 6 hours posttreatment and for (G) cell viability measured by TMRM staining-flow cytometry 6 and 48 hpt. Data are the means ± SD of 3 independent experiments (*P < .05; **P < .01; ***P < .001).
Combinatorial treatment with CFZ and AGI-6780 acts through the inhibition of the NAMPT/SIRT/IDH2 pathway. (A) Schematic representation of the NAMPT/SIRT3/IDH2 pathway and inhibitors. (B) KMS-27 cells treated with DMSO, AGI-6780 (5 µM), CFZ (3 nM), or a combination of the 2 drugs were analyzed for NF-κB activity 6 hours posttreatment. NF-κB activity was detected in total extracts measuring the DNA-binding capability of NF-κB on its target sequence (see “Materials and methods”). Data represent the percentage of NF-κB binding activity normalized vs DMSO samples and are the means ± SD of 3 independent experiments. (C) KMS-27 cells UT, treated with DMSO, CFZ (2.5 nM), AGI-6780 (5 µM), or a combination of the 2 drugs were analyzed for NAMPT mRNA expression levels 24 hours posttreatment. Data are the means ± SD of 3 independent experiments. (D-E) KMS-27 cells were left UT, treated with DMSO or FK866 (10 nM), for 48 hours; vehicle or CFZ (2.5 nM) were added for additional 48 hours. Cells were analyzed for (D) IDH2 activity 6 hours posttreatment with CFZ and for (E) cell viability by TMRM staining-flow cytometry 6 and 48 hpt with CFZ. Data are the means ± SD of 3 independent experiments. (F-G) KMS-27 cells UT, treated with DMSO, 1.25 nM CFZ, 10 µM AGK7, or a combination of the 2 drugs were analyzed for (F) IDH2 activity 6 hours posttreatment and for (G) cell viability measured by TMRM staining-flow cytometry 6 and 48 hpt. Data are the means ± SD of 3 independent experiments (*P < .05; **P < .01; ***P < .001).
Targeting IDH2 and proteasome activities triggers synergistic inhibition of human MM cells growth ex vivo and in vivo with low toxicity to normal human cells
To evaluate whether IDH2 inhibition potentiates CFZ effect in primary cells from MM patients, buffy coats derived from bone marrow aspirates of 9 MM patients were cultured on a layer of HS-5, a bone marrow stromal cell line. Ex vivo cocultures were treated with the CFZ/AGI-6780 combination or with the single drugs for 96 hours. Combination treatment significantly decreased viability of CD138+ cells (Figure 6A). Next, we demonstrated that CFZ/AGI-6780 treatment exhibited a favorable cytotoxicity profile toward peripheral blood mononuclear cells and bone marrow–derived stromal cells compared with KMS-27 (Figure 6B-C). Taking into account that AGI-6780 is not suitable for in vivo studies38 and that enasidenib (AG-221), the mutant IDH2 inhibitor used in the clinic, does not affect the activity of wild-type IDH2,21 we exploited a conditional RNA interference method to knock down IDH2 expression.39,40 To provide an in vivo proof of principle that IDH2 inhibition could increase the therapeutic efficacy of PI in MM, we expressed an IDH2-shRNA (IDH2-A4) in KMS-27 cell line under the control of the DOXY-regulated transcriptional repressor TK. We next studied the growth patterns of KMS-27-TK-IDH2-A4 cells injected subcutaneously into the flanks of NOD/SCID/IL2Rγ−/− (NSG) mice. Mice with masses of 0.5 cm in diameter were treated with DOXY (0.1 mg/mL biweekly), CFZ (4 mg/kg biweekly), or control diluents. Administration of either agent had a substantial effect on tumor growth compared with control mice (P < .0001). Importantly, when IDH2 silencing was combined with CFZ, there was a further significant reduction in tumor growth in relation to single treatments (CFZ vs CFZ/DOXY, P = .0244; DOXY vs CFZ/DOXY, P = .0238; Figure 6D). The median overall survival of mice treated with CFZ associated with IDH2 silencing was significantly longer than vehicle-treated mice (26 vs 49 days; P = .0001) or mice treated with either drug alone (35 days for CFZ and 38 days for DOXY) (Figure 6E). Together, these findings indicate that the antitumor activity of CFZ/AGI-6780 combination extends to primary explants from MM patients with a favorable therapeutic index and provide an in vivo proof of principle that IDH2 inhibition could increase the therapeutic efficacy of PI.
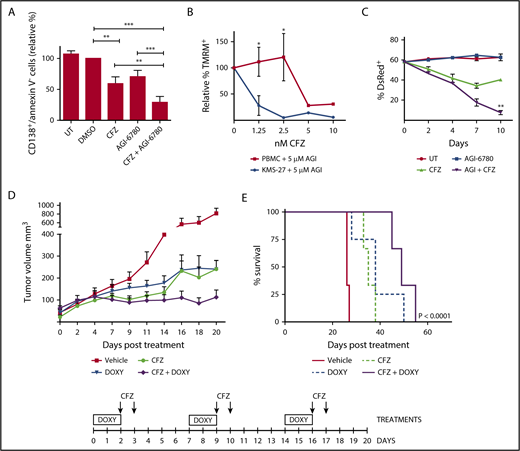
Targeting IDH2 and proteasome activities triggers synergistic inhibition of human MM cells growth ex vivo and in vivo with low toxicity to normal human cells. (A) Buffy coats derived from bone marrow aspirates of MM patients were treated with CFZ (2.5 nM) in combination or not with AGI-6780 (5 μM). Cell viability was estimated by flow cytometry measuring annexin V− and CD138+ cells 96 hours posttreatment. Histograms represent the percentage of viable cells normalized vs DMSO samples. Data are the means ± SEM of 9 independent MM patients. (B) PBMC and KMS-27 were treated with AGI-6780 and increasing doses of CFZ. PBMC were derived from 4 healthy donors. Cell viability was measured by TMRM staining-flow cytometry 48 hours posttreatment. Data are the means ± SD (*P < .05; **P < .01; ***P < .001). (C) KMS-27-TK cells (expressing DsRed fluorescent protein) were co-cultured with HS-5 bone marrow/stroma cell line and treated with CFZ, AGI-6780, or a combination. Percentage of live DsRed+ cells was measured overtime. Data are the means ± SD of 3 independent experiments (CFZ vs CFZ+AGI-6780, **P < .01). (D) Growth patterns of KMS-27-TK-IDH2-A4 cells injected subcutaneously into the flanks of NSG mice. Tumor masses of 0.5 cm diameter mice were randomized for treatment with vehicle (n = 6), 4 mg/kg CFZ (n = 8), 0.1 mg/mL DOXY (n = 10), or a combination of both compounds (n = 10) over 3 weeks. Administration of either agent had a substantial effect on tumor growth compared with control mice (P < .0001). Combination of IDH2 silencing with CFZ further reduced tumor growth in relation to single treatments (CFZ vs CFZ/DOXY, P = .0244; DOXY vs CFZ/DOXY, P = .0238). Each data point represents the average tumor volume (mean ± SEM) for the indicated treatment condition. The timeline shows the schedule of treatment followed for in vivo treatments. (E) Kaplan-Meier survival plot showing survival for mice treated with vehicle (n = 6), 4 mg/kg CFZ (n = 6), 0.1 mg/mL DOXY (n = 8), or a combination (n = 6). CFZ plus DOXY-treated mice show significantly increased survival (49 days) in comparison with vehicle-treated mice (26 days; P < .0001), CFZ alone (35 days; P = .0007), and DOXY alone (38 days; P = .0472). PBMC, Peripheral blood mononuclear cell; SEM, standard error of the mean.
Targeting IDH2 and proteasome activities triggers synergistic inhibition of human MM cells growth ex vivo and in vivo with low toxicity to normal human cells. (A) Buffy coats derived from bone marrow aspirates of MM patients were treated with CFZ (2.5 nM) in combination or not with AGI-6780 (5 μM). Cell viability was estimated by flow cytometry measuring annexin V− and CD138+ cells 96 hours posttreatment. Histograms represent the percentage of viable cells normalized vs DMSO samples. Data are the means ± SEM of 9 independent MM patients. (B) PBMC and KMS-27 were treated with AGI-6780 and increasing doses of CFZ. PBMC were derived from 4 healthy donors. Cell viability was measured by TMRM staining-flow cytometry 48 hours posttreatment. Data are the means ± SD (*P < .05; **P < .01; ***P < .001). (C) KMS-27-TK cells (expressing DsRed fluorescent protein) were co-cultured with HS-5 bone marrow/stroma cell line and treated with CFZ, AGI-6780, or a combination. Percentage of live DsRed+ cells was measured overtime. Data are the means ± SD of 3 independent experiments (CFZ vs CFZ+AGI-6780, **P < .01). (D) Growth patterns of KMS-27-TK-IDH2-A4 cells injected subcutaneously into the flanks of NSG mice. Tumor masses of 0.5 cm diameter mice were randomized for treatment with vehicle (n = 6), 4 mg/kg CFZ (n = 8), 0.1 mg/mL DOXY (n = 10), or a combination of both compounds (n = 10) over 3 weeks. Administration of either agent had a substantial effect on tumor growth compared with control mice (P < .0001). Combination of IDH2 silencing with CFZ further reduced tumor growth in relation to single treatments (CFZ vs CFZ/DOXY, P = .0244; DOXY vs CFZ/DOXY, P = .0238). Each data point represents the average tumor volume (mean ± SEM) for the indicated treatment condition. The timeline shows the schedule of treatment followed for in vivo treatments. (E) Kaplan-Meier survival plot showing survival for mice treated with vehicle (n = 6), 4 mg/kg CFZ (n = 6), 0.1 mg/mL DOXY (n = 8), or a combination (n = 6). CFZ plus DOXY-treated mice show significantly increased survival (49 days) in comparison with vehicle-treated mice (26 days; P < .0001), CFZ alone (35 days; P = .0007), and DOXY alone (38 days; P = .0472). PBMC, Peripheral blood mononuclear cell; SEM, standard error of the mean.
Discussion
Even though PI have led to substantial outcome improvements in MM and MCL patients, development of novel combination strategies is needed to overcome resistance and broaden the applicability of this class of drugs to other malignancies.
The present study identified IDH2 as a new synthetic lethal target to PI, efficacious in several hematological malignancies including MM, MCL, and BL. We showed that the combined targeting of IDH2 and proteasome triggers synergistic inhibition of human MM ex vivo and in vivo, with low toxicity to normal human cells. We demonstrated that the NAMPT/SIRT3/IDH2 pathway is a major determinant of PI responsiveness, thus providing a proof of concept for new combination strategies to enhance sensitivity and broaden the application of PI to other malignancies.
IDH2 is a mitochondrial enzyme that catalyzes the reversible oxidative decarboxylation of isocitrate to α-ketoglutarate, with concomitant reduction of NADP+ to NADPH. Hotspot mutations in IDH2 gene have been identified in acute myeloid leukemia (AML),41,42 angioimmunoblastic T-cell lymphoma,43 and several other malignancies.42,44-47 IDH2 mutations cause a loss of IDH2 activity and an enzymatic gain of function that catalyzes the conversion of α-ketoglutarate to (R)-hydroxyglutarate, with consequences on metabolism, epigenetic state, and cellular differentiation.48,49 The appreciation of the role of IDH2 mutations in oncogenesis and their early occurrence prompted the approval of the IDH2-mutant inhibitor enasidenib (AG-221) for treating refractory/relapsed IDH2-mutated AML patients.
In contrast, the potential role of wild-type IDH2 and its clinical relevance in cancers has been poorly investigated. It is thought that the effect of IDH2 expression on neoplastic progression and drug resistance differs with respect to the site of origin and histological type.50-55 Our study suggests the hypothesis that inhibition of wild-type IDH2 may have therapeutic potentials, regardless of IDH2 expression levels. Concordantly, we excluded that the IDH2 mutational status or its aberrant expression was associated to PI responsiveness in MM cells. Analysis of gene expression profiling datasets did not detect significant changes of IDH2 expression in the evolution of MM disease (data not shown). However, evaluation of IDH2 enzymatic activity could be more appropriate to further dissect the relevance of IDH2 in tumor development and maintenance, as well as a possible prognostic factor.
We demonstrated that genetic and pharmacological inhibition of IDH2 synergizes with first- and second-generation PI by enhancing tumor cells death. In contrast, induction of IDH2 enzymatic activity through glucose starvation impairs the therapeutic efficacy of PI, confirming that pharmacological IDH2 inhibition is a suitable strategy to enhance PI effects.
Mechanistically, we observed that CFZ significantly downregulates NAMPT expression levels, most likely through the inhibition of NF-κB.33-35 NAMPT is a key NAD pathway intermediate that catalyzes the transfer of a phosphoribosyl group from 5-phosphoribosyl-1-pyrophosphate to nicotinamide, forming nicotinamide mononucleotide. It has been previously shown that NAMPT inhibition is synthetic lethal to BTZ in MM.36 Here, we demonstrated that combination of CFZ with either NAMPT or SIRT3 inhibitors induces synergistic downregulation of IDH2 activity through the impairment of NAMPT/SIRT3/IDH2 pathway. The strong impairment of this pathway drastically decreases IDH2 and TCA cycle activities, leading to ETC flux and mitochondrial ATP synthesis downregulation. However, we could not exclude that additional mechanisms may contribute to the antitumoral effects of CFZ/AGI-6780 combination.
We showed that the combined targeting of IDH2 and proteasome activities triggers synergistic inhibition of primary human MM cells. Importantly, CFZ/AGI-6780 combination exhibits a favorable cytotoxicity profile toward peripheral blood mononuclear cells and bone marrow–derived stromal cells. Considering the efficacy of CFZ/AGI-6780 in PI-resistant cell lines as well, we speculate that this combination could be successful in relapsed and refractory MM patients. To answer this question, further studies in a cohort of patients stratified for their PI response are required. We extended the clinical relevance of our observations proving that IDH2 inhibition synergizes with PI in several B-cell non-Hodgkin lymphoma cell lines including MM, MCL, BL, and diffuse large B-cell lymphomas (data not shown). Our preclinical studies therefore provide the rationale for development of novel IDH2 inhibitors directed against wild-type IDH2. These observations are in line with recent studies highlighting the importance of wild-type IDH1 as therapeutic potential.56-58 A further interesting expansion to the present work would be to investigate whether IDH2 synthetic lethal interaction to PI could also take place in cancer patients with mutant IDH2, such as AML, angioimmunoblastic T-cell lymphoma, and other malignancies.
Finally, we provided an in vivo proof of principle that IDH2 inhibition enhances the therapeutic efficacy of CFZ in a subcutaneous xenograft model of MM, resulting in inhibition of tumor progression and extended survival. Owing to the lack of wild-type IDH2 inhibitors suitable for an vivo use, we exploited a conditional shRNA system to knock down IDH2. In contrast to in vitro data, in vivo IDH2 inhibition has a more substantial effect on tumor growth, probably as a consequence of higher gene silencing obtained with the inducible shRNA.
In conclusion, the present study identified IDH2 as a new synthetic lethal target to PI that is efficacious in several hematological malignancies. We demonstrated that the NAMPT/SIRT3/IDH2 pathway is a major determinant of PI responsiveness, providing a proof of concept for new combination strategies to enhance sensitivity and broaden the application of PI to other malignancies.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported by grants from the Associazione Italiana per la Ricerca sul Cancro, Milano, Italy (R.P. and A.N.); Fondazione CRT, Torino, Italy (R.P.); University of Torino (R.P.), and the Gelu Foundation (F.B.).
Authorship
Contribution: E.B. carried out most of the experiments and contributed to the interpretation of biological data with G.G., E.M., E.P., N.V., C.B., V.P., S.D., and F.B.; G.G. and E.M. performed short hairpin RNA screening experiments and analysis; C.R. and V.A. performed biochemical studies. N.V. performed tumor xenograft studies; A. Ricci and A. Rossi developed proteasome inhibitor–resistant cell lines; K.T., L.C., and A.N. performed gene expression profiling experiments and bioinformatics analyses; P.O. and A.P. provided clinically annotated multiple myeloma samples; R.P. designed the studies and supervised the project; R.P. and E.B. wrote the manuscript; and all authors were involved in the final version of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Roberto Piva, Department of Molecular Biotechnology and Health Sciences, University of Torino, via Nizza 52, Torino, 10126 Italy; e-mail: roberto.piva@unito.it.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal