

In this issue of Blood, report the presence of functional redundancy between the transcription factors lymphoblastic leukemia 1 protein (LYL1) and stem cell leukemia protein (SCL) in the process of megakaryocyte (MK) maturation and platelet production in mice.1 This discovery may provide an answer to the long-standing mystery of why mutations in SCL protein do not present with any phenotypic platelet disorder, whereas mutations in its binding partner proteins GATA1, FLI1, RUNX1, and GFI1B cause hereditary thrombocytopenia.
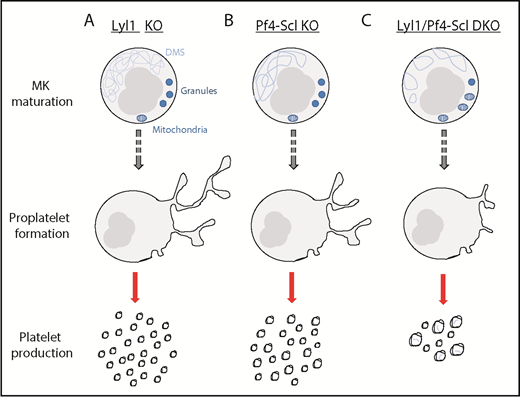
The effect of Lyl1-KO, Pf4-Scl-KO, and Lyl1/Pf4-Scl-DKO on megakaryocyte maturation, proplatelet formation, and platelet production. (A) Lyl1-KO mice presented with a phenotype similar to that of wild-type mice. (B) Pf4-Scl-KO mice exhibited mild phenotypic differences, including a less well-developed DMS and a slight macrothrombocytopenia. (C) Lyl1/Pf4-Scl-DKO mice presented with a severe phenotype, including an abnormal vesicular DMS, reduced number of granules, prominent mitochondria, reduced proplatelet formation, and marked macrothrombocytopenia with the resulting platelets containing DMS remnants.
The effect of Lyl1-KO, Pf4-Scl-KO, and Lyl1/Pf4-Scl-DKO on megakaryocyte maturation, proplatelet formation, and platelet production. (A) Lyl1-KO mice presented with a phenotype similar to that of wild-type mice. (B) Pf4-Scl-KO mice exhibited mild phenotypic differences, including a less well-developed DMS and a slight macrothrombocytopenia. (C) Lyl1/Pf4-Scl-DKO mice presented with a severe phenotype, including an abnormal vesicular DMS, reduced number of granules, prominent mitochondria, reduced proplatelet formation, and marked macrothrombocytopenia with the resulting platelets containing DMS remnants.
Platelet production is an elaborate and complex process that begins with the differentiation of platelet precursor cells (MKs) from hematopoietic stem cells (HSCs). In brief, HSCs are predominately found in the bone marrow, where they become lineage committed and mature into MKs.2,3 After maturation, MKs use their cytoskeleton to reorganize their cytoplasm and its contents into proplatelets, which are released into the bloodstream.2,3 HSC fate and MK differentiation are largely coordinated by the temporal expression of various transcription factors under the direction of thrombopoietin. A substantive body of literature has identified the transcription factors SCL, GATA1, FLI1, RUNX1, NFE2, and GFI1B as key regulators of MK maturation and platelet formation.4 Likewise, the loss of expression of many of these transcription factors in mice results in severe consequences; Nfe2 knockout (KO) mice lack circulating platelets,5 and Gata1-deficient mice die before birth from severe anemia.6 However, mutations in the transcription factor SCL surprisingly do not result in thrombocytopenia. Chiu and colleagues hypothesized that this was not because it plays a nonessential role, but rather because there is a functional redundancy between SCL and a highly related basic helix-loop-helix factor LYL1.
Previous studies have been unable to address the dual roles of SCL and LYL1 because of embryonic lethality associated with the constitutive deletion of Scl.7 To circumvent this, the authors used platelet-specific Pf4Cre to delete Scl in mice with an Lyl1-null background, thus enabling the study of megakaryopoiesis in megakaryocytes lacking both Scl and Lyl1. In whole blood, Pf4Sclc-KO mice exhibited mild macrothrombocytopenia compared with wild-type (WT) littermate controls, consistent with a previous study,8 whereas Lyl1-KO mice had no abnormal phenotype. Interestingly, double knockout (DKO) mice displayed a severe macrothrombocytopenia (20% of WT). See figure for schematic detailing the differences between phenotypes. Striking transmission electron micrographs showed that DKO megakaryocytes had abnormal morphology, including a misformed demarcation membrane system (DMS), the membrane system within MKs thought to provide the plasma membrane for future platelets, markedly reduced α-granules, and prominent mitochondria. These DKO MKs were also approximately twice as abundant within the bone marrow. In addition, DKO platelets contained small rounded vesicles and fragments of DMSs, both highly unusual in normal platelets. The hemostatic consequences of the absence of Scl and Lyl1 was seen in a significantly prolonged bleeding time in DKO compared with single-KO mice. In addition, both Pf4Sclc and the DKO platelets showed reduced P-selectin expression in response to thrombin, which indicated not only a defect in platelet formation from MKs but also a functional platelet defect.
To pinpoint exactly which genes SCL and LYL1 were targeting, the authors performed RNA sequencing on MKs cultured from each mouse genotype. Although there were 384 and 40 genes differentially expressed in the Pf4Sclc and Lyl1 KO MKs, respectively, 1963 genes were differentially expressed in the DKO mice, including Gata1, Fli1, and Nfe2 and genes responsible for platelet activation. Subsequent chromatin immunoprecipitation analysis further confirmed that SCL and LYL1 both target partner transcription factors GATA1 and NFE2. This result is of particular interest because many of the observations made in the DKO mice phenocopy Nfe2 KO mice, including the excess of bone marrow MKs, misformed DMSs, reduced granules, and inability to efficiently shed platelets into the bloodstream.9,10 Unlike the Nfe2 KO MKs, however, the Scl and Lyl1 DKO MKs also had a defect in proplatelet formation in vitro, suggesting that the phenotype goes beyond regulation of Nfe2 alone. Future studies interrogating the reason for these additional defects will undoubtedly be important.
In sum, the use of the MK-specific Pf4Cre model allowed Chiu and colleagues to definitively examine megakaryopoiesis in the absence of both Scl and Lyl1, thus determining that both genes are important and are able to compensate for each other’s absence, explaining why SCL has not been associated with thrombocytopenia. This study underscores the significance of letting human pathology guide lines of inquiry and also highlights the utility of murine models in answering those questions.
Conflict-of-interest disclosure: The authors declare no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal